The hardware and bandwidth for this mirror is donated by dogado GmbH, the Webhosting and Full Service-Cloud Provider. Check out our Wordpress Tutorial.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]dogado.de.
![]()
![]()
The goal of yatah is to manage taxonomy when
lineages are described with strings and ranks separated with special
patterns like |*__ or ;*__.
For instance, the well-known Escherichia coli could be coded
as
k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Enterobacteriales|f__Enterobacteriaceae|g__Escherichia|s__Escherichia_coli.
You can install the released version of yatah from CRAN with:
install.packages("yatah")And the development version from GitHub with:
# install.packages("remotes")
remotes::install_github("abichat/yatah")library(yatah)yatah handles 8 different ranks:
all_ranks
#> [1] "kingdom" "phylum" "class" "order" "family" "genus" "species" "strain"A lineage is composed of a succession of clades separated by special characters indicating the current rank.
lineages <- c(
"k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Coriobacteriales",
"k__Bacteria|p__Bacteroidetes|c__Bacteroidia|o__Bacteroidales",
"k__Bacteria|p__Bacteroidetes|c__Flavobacteriia|o__Flavobacteriales",
"k__Bacteria|p__Firmicutes|c__Bacilli|o__Bacillales",
"k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales",
"k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales",
"k__Bacteria|p__Proteobacteria|c__Epsilonproteobacteria|o__Campylobacterales",
"k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Enterobacteriales",
"k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Pseudomonadales"
)yatah offers functions to verify if lineages meet a desired property, to extract information, or to compute summary objects.
is_rank() and is_at_least_rank() check if
the lineages are of the desired rank.is_rank(lineages, rank = "order")
#> [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
is_at_least_rank(lineages, rank = "species")
#> [1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSEis_clade() checks if the lineages belong to the desired
clade.is_clade(lineages, clade = "Proteobacteria", rank = "phylum")
#> [1] FALSE FALSE FALSE FALSE FALSE FALSE TRUE TRUE TRUEget_clade() extracts the clade of the desired
rank.get_clade(lineages, rank = "class")
#> [1] "Actinobacteria" "Bacteroidia" "Flavobacteriia" "Bacilli"
#> [5] "Bacilli" "Clostridia" "Epsilonproteobacteria" "Gammaproteobacteria"
#> [9] "Gammaproteobacteria"get_last_clade() extracts the last clade of the
lineages.get_last_clade(lineages)
#> [1] "Coriobacteriales" "Bacteroidales" "Flavobacteriales" "Bacillales" "Lactobacillales"
#> [6] "Clostridiales" "Campylobacterales" "Enterobacteriales" "Pseudomonadales"get_all_clades() extracts all clades of the
lineages.get_all_clades(lineages, simplify = TRUE)
#> [1] "Actinobacteria" "Bacillales" "Bacilli" "Bacteria"
#> [5] "Bacteroidales" "Bacteroidetes" "Bacteroidia" "Campylobacterales"
#> [9] "Clostridia" "Clostridiales" "Coriobacteriales" "Enterobacteriales"
#> [13] "Epsilonproteobacteria" "Firmicutes" "Flavobacteriales" "Flavobacteriia"
#> [17] "Gammaproteobacteria" "Lactobacillales" "Proteobacteria" "Pseudomonadales"taxtable() computes the taxonomic table corresponding
to the lineages.table <- taxtable(lineages)
table
#> kingdom phylum class order
#> 1 Bacteria Actinobacteria Actinobacteria Coriobacteriales
#> 2 Bacteria Bacteroidetes Bacteroidia Bacteroidales
#> 3 Bacteria Bacteroidetes Flavobacteriia Flavobacteriales
#> 4 Bacteria Firmicutes Bacilli Bacillales
#> 5 Bacteria Firmicutes Bacilli Lactobacillales
#> 6 Bacteria Firmicutes Clostridia Clostridiales
#> 7 Bacteria Proteobacteria Epsilonproteobacteria Campylobacterales
#> 8 Bacteria Proteobacteria Gammaproteobacteria Enterobacteriales
#> 9 Bacteria Proteobacteria Gammaproteobacteria Pseudomonadalestaxtree() computes the taxonomic tree (format
phylo) from a taxonomic table.tree <- taxtree(table)
tree
#>
#> Phylogenetic tree with 9 tips and 6 internal nodes.
#>
#> Tip labels:
#> Coriobacteriales, Bacteroidales, Flavobacteriales, Bacillales, Lactobacillales, Clostridiales, ...
#> Node labels:
#> Bacteria, Bacteroidetes, Firmicutes, Bacilli, Proteobacteria, Gammaproteobacteria
#>
#> Rooted; includes branch lengths.
plot(tree, show.node.label = TRUE)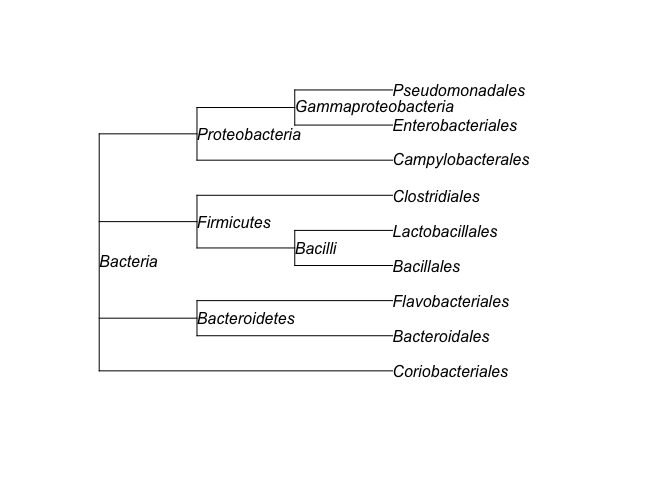
If you want to change the default separator from | to,
e.g., ;, use options(yatah_sep = ";"). Reset
it with options(yatah_sep = "\\|").
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
Health stats visible at Monitor.