
The hardware and bandwidth for this mirror is donated by dogado GmbH, the Webhosting and Full Service-Cloud Provider. Check out our Wordpress Tutorial.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]dogado.de.
![]()
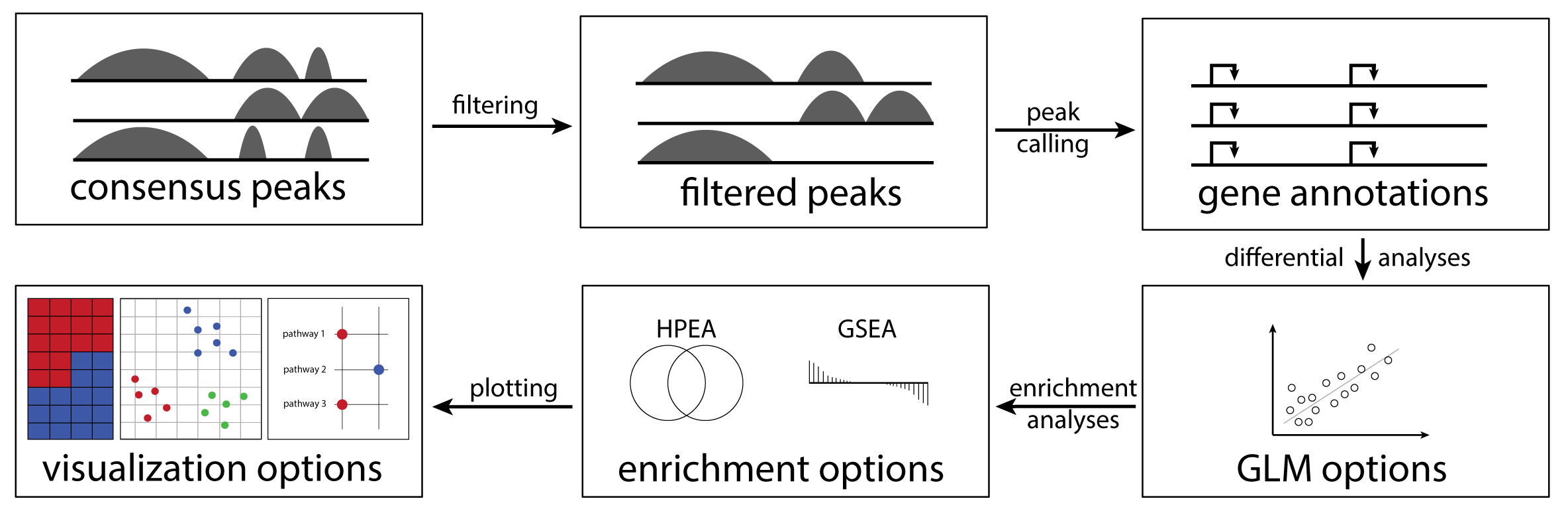
cinaR is a single wrapper function for end-to-end
computational analyses of bulk ATAC-seq (or RNA-seq) profiles. Starting
from a consensus peak file, it outputs differentially accessible peaks,
enrichment results, and provides users with various configurable
visualization options. For more details, please see the preprint.
# CRAN mirror
install.packages("cinaR")To get bug fix and use a feature from the development version:
# install.packages("devtools")
devtools::install_github("eonurk/cinaR")Sometimes bioconductor related packages may not be installed
automatically.
Therefore, you may need to install them manually:
BiocManager::install(c("ChIPseeker", "DESeq2", "edgeR", "fgsea","GenomicRanges", "limma", "preprocessCore", "sva", "TxDb.Hsapiens.UCSC.hg38.knownGene", "TxDb.Hsapiens.UCSC.hg19.knownGene", "TxDb.Mmusculus.UCSC.mm10.knownGene"))library(cinaR)
#> Checking for required Bioconductor packages...
#> All required Bioconductor packages are already installed.
# create contrast vector which will be compared.
contrasts<- c("B6", "B6", "B6", "B6", "B6", "NZO", "NZO", "NZO", "NZO", "NZO", "NZO",
"B6", "B6", "B6", "B6", "B6", "NZO", "NZO", "NZO", "NZO", "NZO", "NZO")
# If reference genome is not set hg38 will be used!
results <- cinaR(bed, contrasts, reference.genome = "mm10")
#> >> Experiment type: ATAC-Seq
#> >> Matrix is filtered!
#>
#> >> preparing features information... 2024-05-22 12:38:01
#> >> identifying nearest features... 2024-05-22 12:38:02
#> >> calculating distance from peak to TSS... 2024-05-22 12:38:02
#> >> assigning genomic annotation... 2024-05-22 12:38:02
#> >> assigning chromosome lengths 2024-05-22 12:38:11
#> >> done... 2024-05-22 12:38:11
#> >> Method: edgeR
#> FDR:0.05& abs(logFC)<0
#> >> Estimating dispersion...
#> >> Fitting GLM...
#> >> DA peaks are found!
#> >> No `geneset` is specified so immune modules (Chaussabel, 2008) will be used!
#> >> enrichment.method` is not selected. Hyper-geometric p-value (HPEA) will be used!
#> >> Mice gene symbols are converted to human symbols!
#> >> Enrichment results are ready...
#> >> Done!
pca_plot(results, contrasts, show.names = F)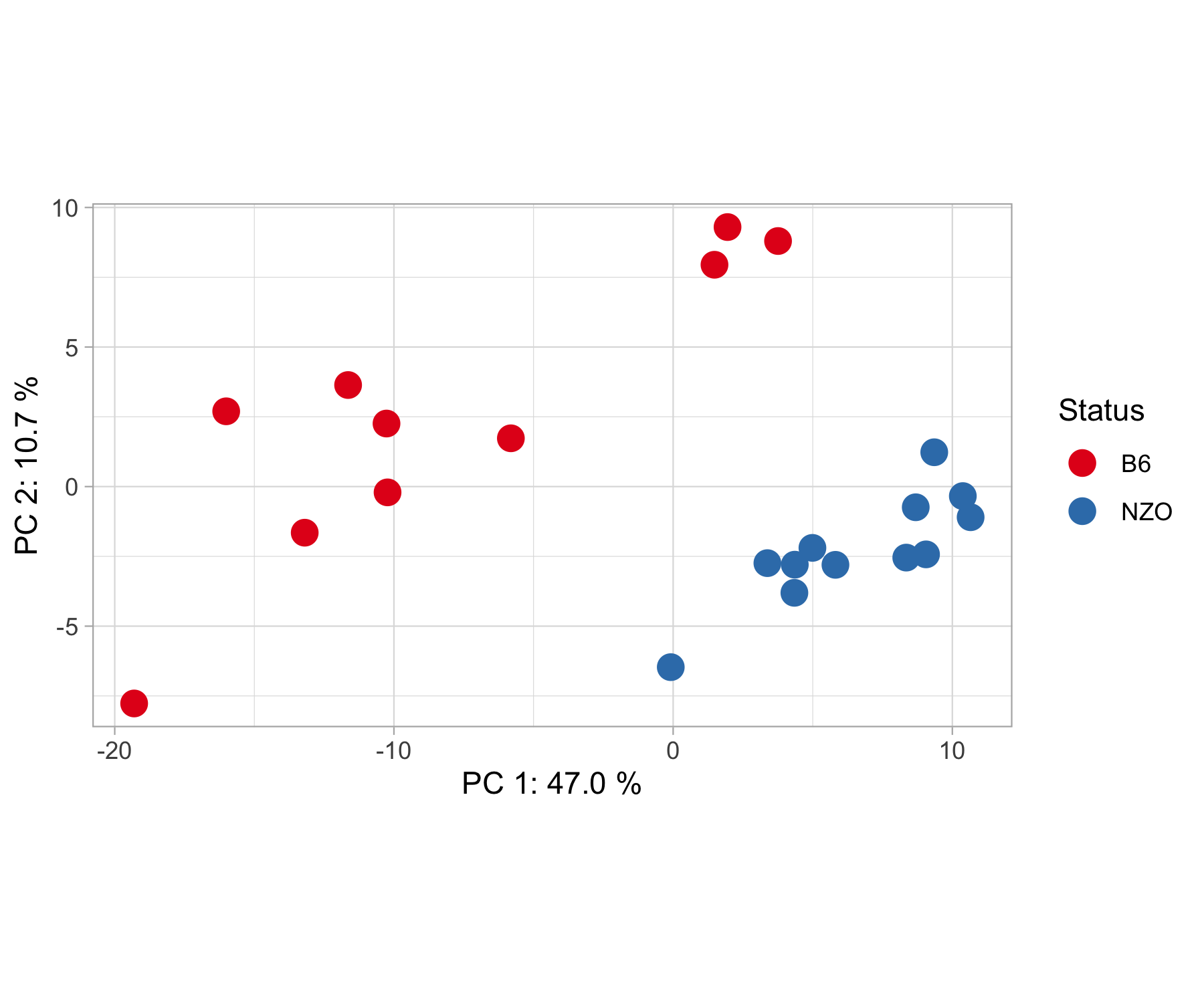
Use prep_scATAC_cinaR() to pseudobulk 10x scATAC
peak-by-cell matrices into a cinaR-ready consensus matrix.
This preserves biological replicates (sample-level) and avoids inflated
significance from per-cell testing.
# counts: peak-by-cell matrix (dense or dgCMatrix)
# meta: data.frame with rownames = cell barcodes
# meta must include biological replicate and condition columns
prep <- prep_scATAC_cinaR(counts, meta,
sample.col = "sample",
group.col = "group")
results <- cinaR(prep$bed, prep$contrasts, reference.genome = "hg38")Per-cell-type (sample × cluster) pseudobulk:
prep_list <- prep_scATAC_cinaR(counts, meta,
sample.col = "sample",
group.col = "group",
cluster.col = "celltype")
results_list <- lapply(prep_list, function(x) {
cinaR(x$bed, x$contrasts, reference.genome = "hg38")
})Seurat/Signac object:
prep <- prep_scATAC_seurat(seurat_obj,
sample.col = "sample",
group.col = "group",
assay = "peaks")
results <- cinaR(prep$bed, prep$contrasts, reference.genome = "hg38")If your peak IDs are not in chr:start-end,
chr_start_end, or chr-start-end format, pass a
peak.bed data.frame with CHR,
START, and STOP columns via
peak.bed = ....
For more details please go to our site from here!
@article {Karakaslar2021.03.05.434143,
author = {Karakaslar, E Onur and Ucar, Duygu},
title = {cinaR: A comprehensive R package for the differential analyses and
functional interpretation of ATAC-seq data},
year = {2021},
doi = {10.1101/2021.03.05.434143},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/10.1101/2021.03.05.434143v2},
journal = {bioRxiv}
}You can send pull requests to make your contributions.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
Health stats visible at Monitor.