The hardware and bandwidth for this mirror is donated by dogado GmbH, the Webhosting and Full Service-Cloud Provider. Check out our Wordpress Tutorial.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]dogado.de.
Bulk sequencing experiments (e.g. mRNAseq, sRNAseq etc) are essential for exploring a wide range of biological questions. To bring the data analysis closer to its interpretation and facilitate both interactive, exploratory tasks and the sharing of (easily accessible) information, we present bulkAnalyseR, an R package that offers a seamless, customisable solution for most bulk sequencing datasets. By integrating state-of-the-art approaches without relying on extensive computational support, and replacing static images with interactive panels, our aim is to further support and strengthen the reusability of data. bulkAnalyseR enables standard analyses of bulk data, using an expression matrix as starting point. It presents the outputs of various steps in an interactive web-based interface, making it easy to generate, explore and verify hypotheses. Moreover, the app can be easily shared and published, incentivising research reproducibility and allowing others to explore the same processed data.
Preprint: https://www.biorxiv.org/content/10.1101/2021.12.23.473982v1
Live app example: https://bioinf.stemcells.cam.ac.uk/shiny/bulkAnalyseR/Yang2019/
If you are using components of this package in published research please cite the following papers along with the bulkAnalyseR manuscript:
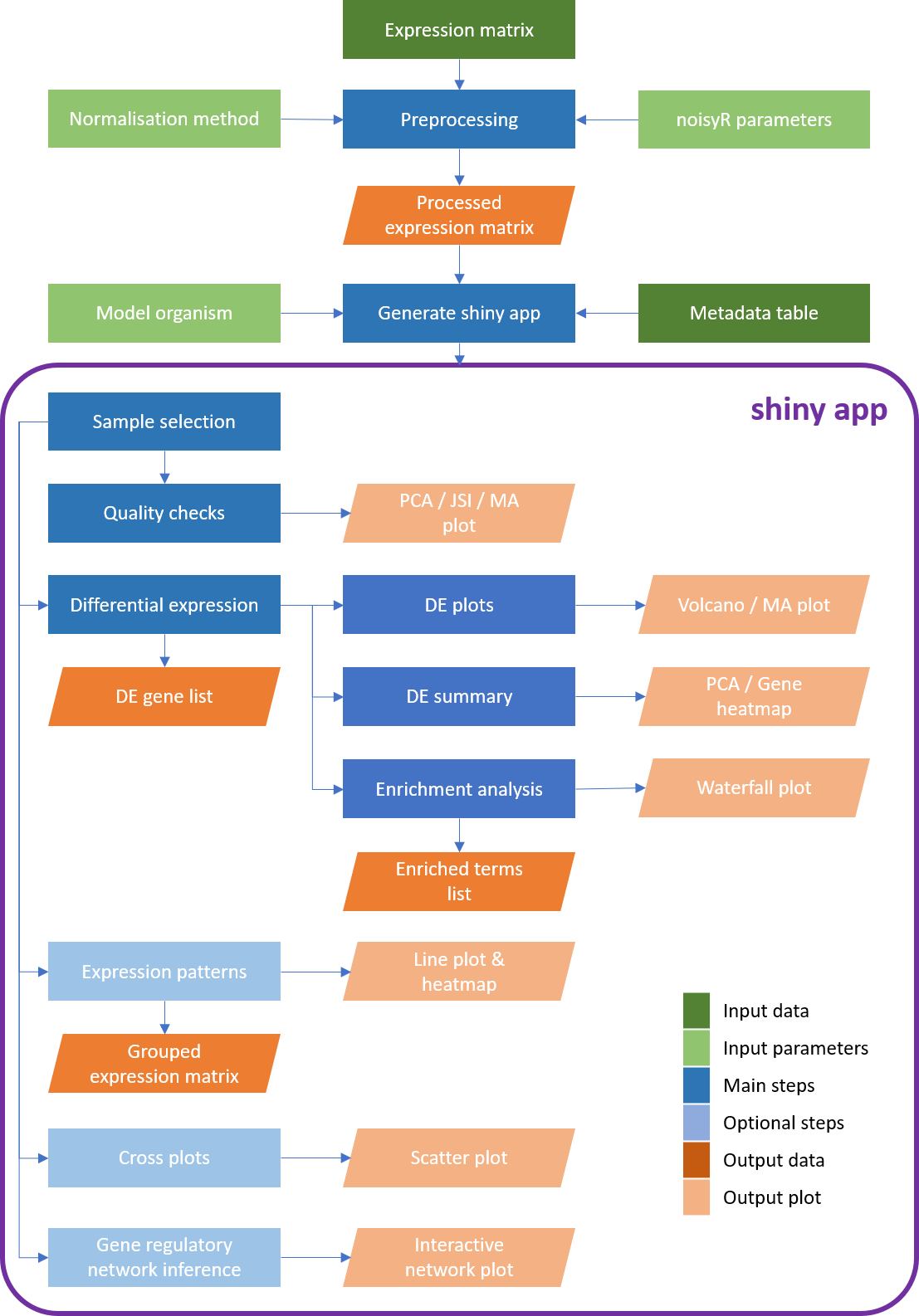
Workflow diagram of the bulkAnalyseR pipeline. The input comprises a processed (i.e. normalised, noise-corrected) expression matrix. Using bulkAnalyseR, all standard steps related to differential expression analyses are handled seamlessley. The pairwaise comparison of differential expression outputs is also possible (using cross plots and upset plots). Finally, localised Gene Regulatory Networks can be created.
To create a shiny app using bulkAnalyseR, you need a processed (e.g. normalised, noise corrected) expression matrix and a corresponding metadata table loaded in your workspace.
The expression matrix is expected to have genes on the rows (with the Ensembl ID as the row name) and samples on the columns.
The first column of the metadata table must match the column names of the expression matrix. The other columns of the metadata table contain other information about the samples e.g. time points, treatment groups, demographic information etc; the differential expression analysis may take into account a subset of these columns.
We illustrate this on a case study from a 2019 paper by Yang et al (https://www.sciencedirect.com/science/article/pii/S2405471219301152). Using three time points, each with two replicates, for some bulk mRNAseq samples, we obtain the following matrices:
| SRR7624365 | SRR7624366 | SRR7624371 | SRR7624372 | SRR7624375 | SRR7624376 | |
|---|---|---|---|---|---|---|
| ENSMUSG00000102693 | 2 | 0 | 0 | 0 | 0 | 0 |
| ENSMUSG00000064842 | 0 | 0 | 0 | 0 | 0 | 0 |
| ENSMUSG00000051951 | 6 | 4 | 2 | 0 | 47 | 37 |
| … | … | … | … | … | … |
Below is an example of a metadata table:
| srr | timepoint |
|---|---|
| SRR7624365 | 0h |
| SRR7624366 | 0h |
| SRR7624371 | 12h |
| SRR7624372 | 12h |
| SRR7624375 | 36h |
| SRR7624376 | 36h |
Before using the expression matrix to create a shiny app, some preprocessing should be performed. bulkAnalyseR contains the function preprocessExpressionMatrix which takes the raw expression matrix as input, then denoises the data using noisyR and normalises the expression levels using either quantile (by default) or RPM normalisation (specified using normalisation.method parameter).
It is not recommended to use data which has not been denoised and normalised as input to generateShinyApp; noisy data is prone to spurious, un-reproducible patterns; analyses performed on unnormalised data are unlikely to be robust. You can also perform your own preprocessing outside preprocessExpressionMatrix function.
Main function: preprocessExpressionMatrix()
Supporting function: noisyr_counts_with_plot()
The central function in bulkAnalyseR is generateShinyApp. This function creates an app.R file and all required objects to run the app in .rda format in the target directory. The key inputs to generateShinyApp are expression.matrix (after being processed using preprocessExpressionMatrix) and meta. You can also specify the title, a folder name where the app will be saved, a shiny theme, as well as specifying the organism on which your data was generated.
Calling generateShinyApp with these parameters will create a folder with your chosen name in which there will be 2 files expression_matrix.rda and metadata.rda and app.R which defines the app. To see the app, you can call shiny::runApp() with the name of the folder as parameter. The app generated is standalone and can be shared with collaborators or published online through a platform like shinyapps.io. This provides an easy way for anyone to explore the data and verify the conclusions, increasing access and promoting reproducibility of the bioinformatics analysis.
By default, the app will have 9 panels: Sample select, Quality checks, Differential expression, Volcano and MA plots, DE summary, Enrichment, Expression patterns, Cross plots, GRN inference. You can choose to remove one or more panels using the default.panels parameter.
By default, the app will look like this:
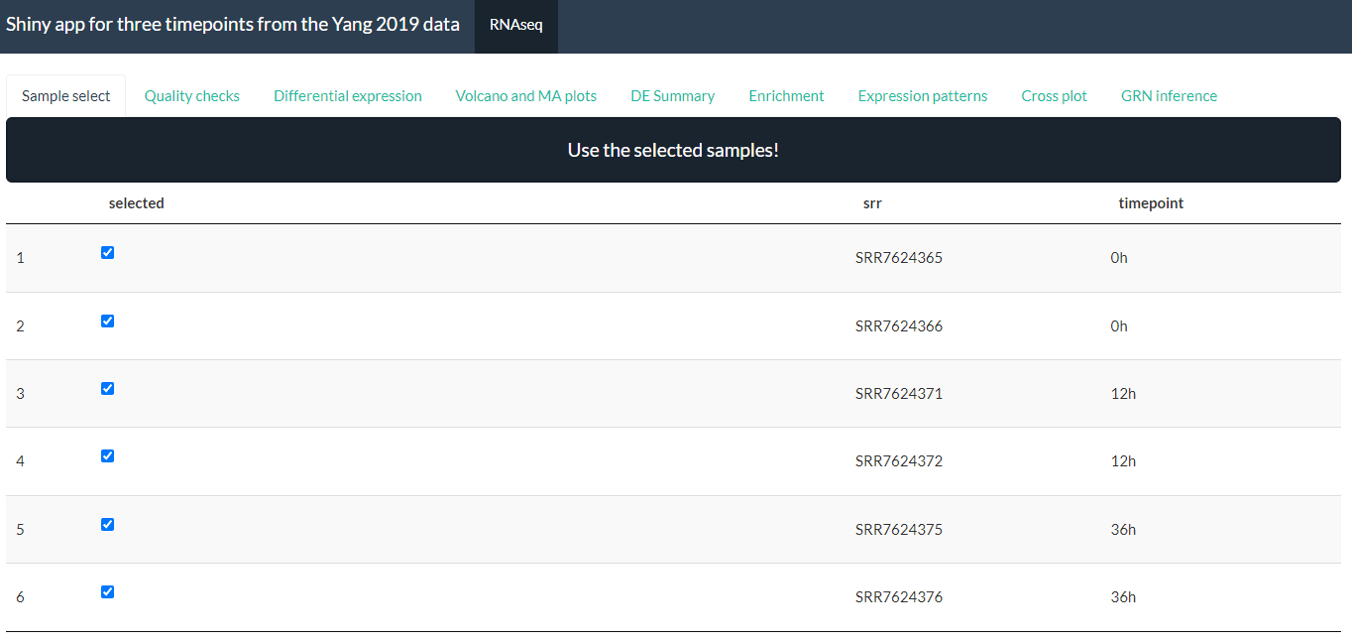
Screenshot from Yang case study processed with the bulkAnalyseR app
See vignette for more details on the individual panels.
You can also add custom extra panels and data using the panels.extra and data.extra parameters.
Main function: generateShinyApp()
Panel functions (with UI and server components): sampleSelectPanel, QCpanel, DEpanel, DEplotPanel, DEsummaryPanel, enrichmentPanel, patternPanel, crossPanel, GRNpanel
Supporting functions: DEanalysis_deseq2, DEanalysis_edger, calculate_condition_mean_sd_per_gene, determine_uds, expression_heatmap, jaccard_heatmap, ma_enhance, ma_plot, make_heatmap_matrix, make_pattern_matrix, plot_line_pattern, plot_pca, volcano_enhance, volcano_plot
A shiny app for the Yang et al 2019 data can be generated using the following code:
library(bulkAnalyseR)
#load expression matrix
expression.matrix <- as.matrix(read.csv(
system.file("extdata", "expression_matrix.csv", package = "bulkAnalyseR"),
row.names = 1
))
#create metadata table
metadata <- data.frame(srr = colnames(expression.matrix),
timepoint = rep(c("0h", "12h", "36h"), each = 2))
#run preprocessing
expression.matrix.preproc <- preprocessExpressionMatrix(expression.matrix)
#create shiny app
shiny.dir <- "shiny_Yang2019"
generateShinyApp(
expression.matrix = expression.matrix.preproc,
metadata = metadata,
shiny.dir = shiny.dir,
app.title = "Shiny app for three timepoints from the Yang 2019 data",
organism = "mmusculus",
org.db = "org.Mm.eg.db"
)
#run shiny app
shiny::runApp(shiny.dir)bulkAnalyseR can be installed from CRAN using install.packages(“bulkAnalyseR”). Please make sure all bioconductor dependencies are also installed.
To install the latest stable development version from GitHub, first install the CRAN dependencies as well as devtools then use devtools::install_github(“Core-Bioinformatics/bulkAnalyseR”).
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
Health stats visible at Monitor.