The hardware and bandwidth for this mirror is donated by dogado GmbH, the Webhosting and Full Service-Cloud Provider. Check out our Wordpress Tutorial.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]dogado.de.
![]()
![]()
![]()
![]()
![]()
CorMID is an R-package providing functions to solve problems during metabolic flux analysis using HR-APCI-MS.
In metabolic flux experiments tracer molecules (often glucose containing labelled carbon) are incorporated in compounds measured using mass spectrometry. The mass isotopologue distributions (MIDs) of these compounds needs to be corrected for natural abundance of labelled carbon and other effects, which are specific on the compound and ionization technique applied. This package provides functions to correct such effects in high resolution gas chromatography atmospheric pressure chemical ionization mass spectrometry (GC-HR-APCI-MS) analyses.
You can install the development version of CorMID from GitHub with:
# install.packages("devtools")
devtools::install_github("janlisec/CorMID")or install the version from CRAN instead.
CorMID is supposed to disentangle a complex MID. Complex means that the ion intensities of the isotopes are influenced by natural abundance, artificial labeling (e.g. by a 13C-Glucose tracer) and mass spectrometry artifacts (i.e. several potential adducts/fragments).
You can create and visualize such a complex mass spectrum, i.e. a vector of measured ion intensities, by providing a chemical formula, the true MID and an adduct distribution like follows:
# a chemical formula, here: Lactic acid 2 TMS
fml <- "C9H22O3Si2"
# the true mass isotopologue distribution, here: 10% U13C enriched
mid <- c(0.9, 0, 0, 0.1)
# adduct distribution, here: 3 different APCI adducts are formed
r <- list("M+H" = 0.8, "M-H" = 0.1, "M+H2O-CH4" = 0.1)
# reconstruct the measured intensity vector
rMID <- CorMID::recMID(mid = mid, r = r, fml = fml)
round(rMID, 3)
#> M-2 M-1 M+0 M+1 M+2 M+3 M+4 M+5
#> 0.069 0.014 0.558 0.121 0.121 0.085 0.018 0.014
#> attr(,"class")
#> [1] "recMID"CorMID provides a class specific plotting function for such a reconstructed MID:
plot(rMID, ylim=c(0,0.6))
mtext(text = "Reconstructed MID", side = 3, line = -1.25, adj = 0.98, outer = T)
text(x = 3, y = 0.4, labels = "[M+H]+", pos=2)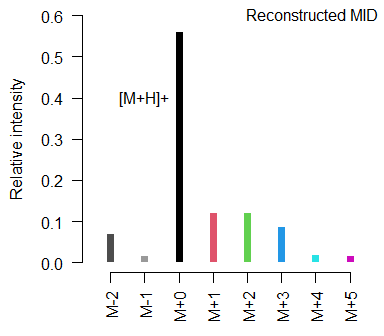
Instead of labeling the mass spectrum with actual ion masses, CorMID unifies the annotation for all molecules with respect to the largest adduct, in APCI usually the [M+H]+, which is labeled as M+0. Other peaks are labeled indicating the approximate mass difference to [M+H]+ in Dalton as M+1, M+2, etc.
Assuming that you have measured the above intensities in your experiment, the main function of CorMID can estimate the underlying MID and r for you:
# disentangle the adduct ratios and true isotopologue distribution (enrichment) from the above test data
out <- CorMID::CorMID(int = rMID, fml = fml)
print(out)
#> [class] 'CorMID'
#> MID [%] (estimated)
#> M0 M1 M2 M3
#> 88.28 00.00 01.56 10.16
#> [attr] 'r' (estimated)
#> M+H M+ M-H M+H2O-CH4
#> 0.81 0.00 0.10 0.09
#> [attr] 'err'
#> 0.003494Please note: no information regarding the true labeling status and the adduct distribution was provided in the above function call. CorMID is able to guess the most likely combination.
This allows you to perform the correction for natural abundance and technical artifacts in a single step and extract the relevant labeling status for flux analysis or other statistical evaluations.
However, in a real world experiment it would be a smart strategy to process non-labeled control samples setting a fixed mid, i.e. c(1, 0, 0, 0) for a molecule with 3 biological carbon atoms like lactic acid, to identify the specific adducts r formed on your device for this molecule. In the next step you can use this information to fix r while processing your labeled samples which will improve the detection of the correct labeling status MID.
You might either read the Vignette describing the package functions in detail or read the publication which shows an evaluation of the performance of CorMID on several real data sets.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
Health stats visible at Monitor.