The hardware and bandwidth for this mirror is donated by dogado GmbH, the Webhosting and Full Service-Cloud Provider. Check out our Wordpress Tutorial.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]dogado.de.
CIDER is a meta-clustering workflow designed to handle scRNA-seq data that span multiple samples or conditions. Often, these datasets are confounded by batch effects or other variables. Many existing batch-removal methods assume near-identical cell population compositions across samples. CIDER, in contrast, leverages inter-group similarity measures to guide clustering without requiring such strict assumptions.
You can install CIDER from github with:
# install.packages("devtools")
devtools::install_github('zhiyuan-hu-lab/CIDER')If you have already integrated your scRNA-seq data (e.g., using Seurat-CCA, Harmony, or Scanorama) and want to evaluate how well the biological populations align post-integration, you can use CIDER as follows.
seu.integrated) with corrected PCs
inseu.integrated@reductions$pca@cell.embeddings`seu.integrated@reductions$pca@cell.embeddings <- corrected.PCslibrary(CIDER)
seu.integrated <- hdbscan.seurat(seu.integrated)
ider <- getIDEr(seu.integrated, verbose = FALSE)
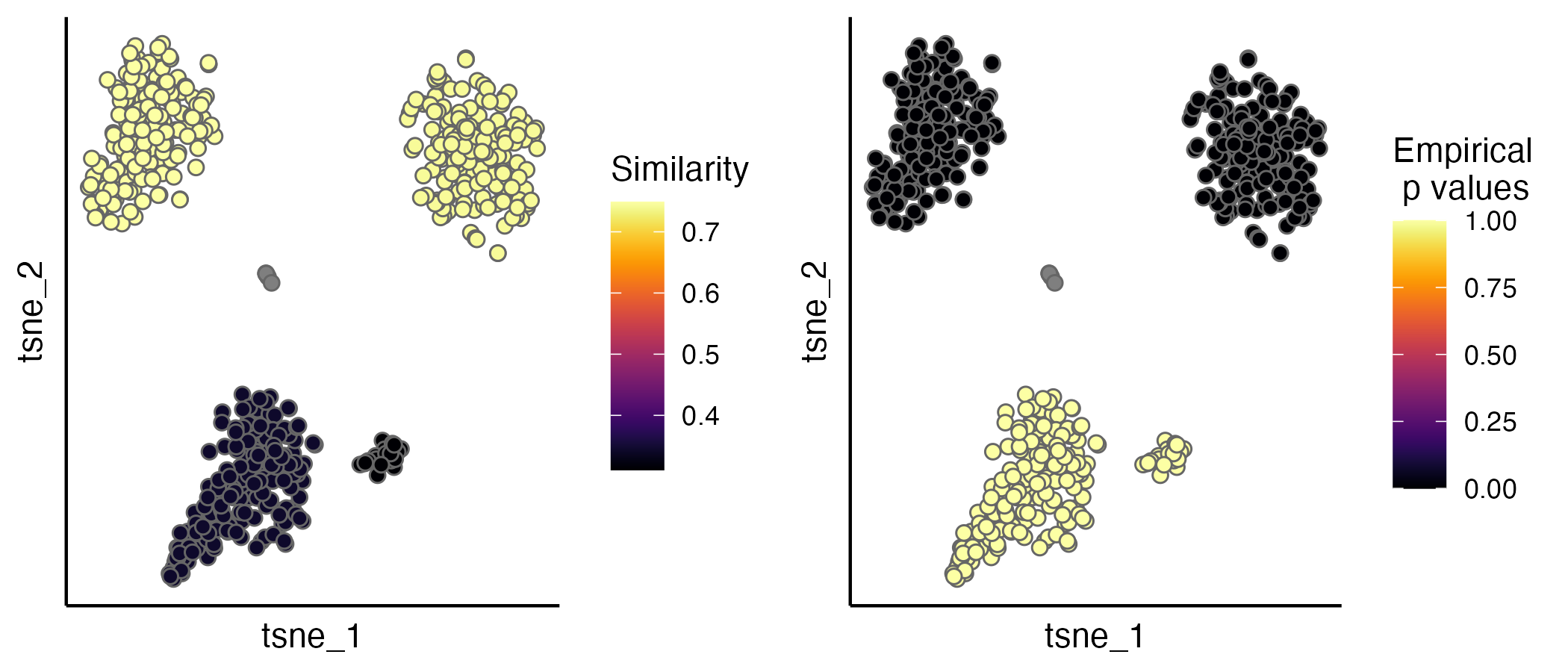
seu.integrated <- estimateProb(seu.integrated, ider)The evaluation scores (IDER-based similarity and empirical p values)
can be visualised by the scatterPlot function.
p1 <- scatterPlot(seu.integrated, "tsne", colour.by = "similarity")
p2 <- scatterPlot(seu.integrated, "tsne", colour.by = "pvalue")
plot_grid(p1,p2, ncol = 2)For a more detailed walkthrough, see the detailed tutorial of evaluation
In many scenarios, you do not start with an integrated Seurat object but still need to cluster multi-batch scRNA-seq data in a robust way. CIDER provides meta-clustering approaches:
If your Seurat object (seu) has:
initial_cluster in seu@meta.data for
per-batch clusters, andBatch for batch labels,then two main steps are:
# Step 1: Compute IDER-based similarity
ider <- getIDEr(seu,
group.by.var = "initial_cluster",
batch.by.var = "Batch")
# Step 2: Perform final clustering
seu <- finalClustering(seu, ider, cutree.h = 0.45)The final clusters will be stored in
seu@meta.data$final_cluster (by default).
If you find CIDER helpful for your research, please cite:
Z. Hu, A. A. Ahmed, C. Yau. CIDER: an interpretable meta-clustering framework for single-cell RNA-seq data integration and evaluation. Genome Biology 22, Article number: 337 (2021); doi: https://doi.org/10.1186/s13059-021-02561-2
Z. Hu, M. Artibani, A. Alsaadi, N. Wietek, M. Morotti, T. Shi, Z. Zhong, L. Santana Gonzalez, S. El-Sahhar, M. KaramiNejadRanjbar, G. Mallett, Y. Feng, K. Masuda, Y. Zheng, K. Chong, S. Damato, S. Dhar, L. Campo, R. Garruto Campanile, V. Rai, D. Maldonado-Perez, S. Jones, V. Cerundolo, T. Sauka-Spengler, C. Yau, A. A. Ahmed. The repertoire of serous ovarian cancer non-genetic heterogeneity revealed by single-cell sequencing of normal fallopian tube epithelial cells. Cancer Cell 37 (2), p226-242.E7 (2020). doi: https://doi.org/10.1101/2021.03.29.437525
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
Health stats visible at Monitor.