The hardware and bandwidth for this mirror is donated by dogado GmbH, the Webhosting and Full Service-Cloud Provider. Check out our Wordpress Tutorial.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]dogado.de.
TOmicsVis: TranscriptOmics Visualization.
Website: https://benben-miao.github.io/TOmicsVis/
New!!! TOmicsVis Shinyapp:
# Start shiny application.
TOmicsVis::tomicsvis()1.2.1 Install required packages from Bioconductor:
# Install required packages from Bioconductor
install.packages("BiocManager")
BiocManager::install(c("ComplexHeatmap", "EnhancedVolcano", "clusterProfiler", "enrichplot", "impute", "preprocessCore", "Mfuzz"))1.2.2 Github: https://github.com/benben-miao/TOmicsVis/
Install from Github:
install.packages("devtools")
devtools::install_github("benben-miao/TOmicsVis")
# Resolve network by GitClone
devtools::install_git("https://gitclone.com/github.com/benben-miao/TOmicsVis.git")1.2.3 CRAN: https://cran.r-project.org/package=TOmicsVis
Install from CRAN:
# Install from CRAN
install.packages("TOmicsVis")Videos Courses: https://space.bilibili.com/34105515/channel/series
Article Introduction: 全解TOmicsVis完美应用于转录组可视化R包
Article Courses: TOmicsVis 转录组学R代码分析及可视化视频
OmicsSuite: Omics Suite Github: https://github.com/omicssuite/
Authors:
# 1. Library TOmicsVis package
library(TOmicsVis)
#> 载入需要的程辑包:Biobase
#> 载入需要的程辑包:BiocGenerics
#>
#> 载入程辑包:'BiocGenerics'
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, aperm, append, as.data.frame, basename, cbind,
#> colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
#> get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
#> match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
#> Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
#> table, tapply, union, unique, unsplit, which.max, which.min
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#> 载入需要的程辑包:e1071
#>
#> Registered S3 method overwritten by 'GGally':
#> method from
#> +.gg ggplot2
#>
#> 载入程辑包:'DynDoc'
#> The following object is masked from 'package:BiocGenerics':
#>
#> path
# 2. Extra package
# install.packages("ggplot2")
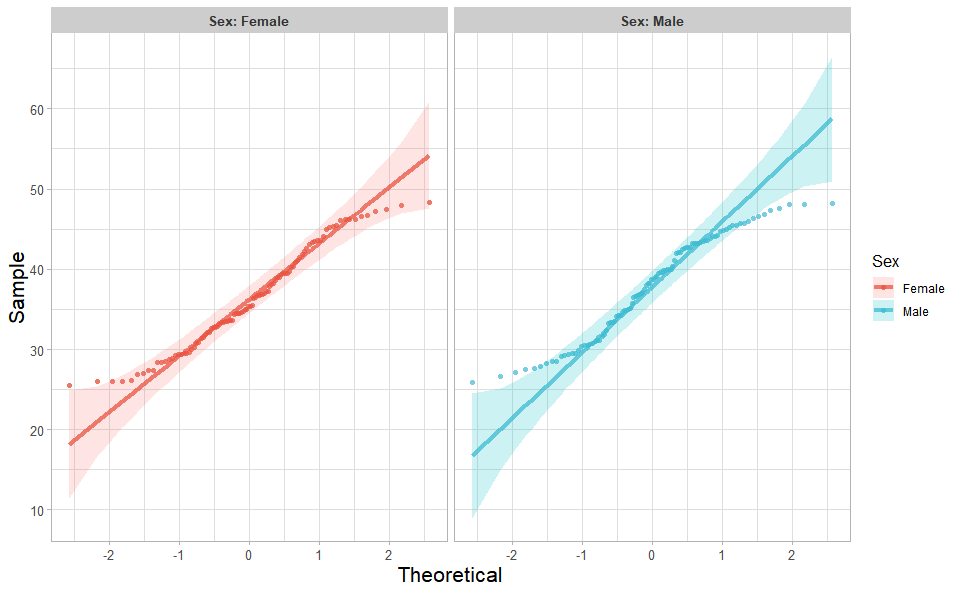
library(ggplot2)Input Data: Dataframe: Weight and Sex traits dataframe (1st-col: Weight, 2nd-col: Sex).
Output Plot: Quantile plot for visualizing data distribution.
# 1. Load example datasets
data(weight_sex)
head(weight_sex)
#> Weight Sex
#> 1 36.74 Female
#> 2 38.54 Female
#> 3 44.91 Female
#> 4 43.53 Female
#> 5 39.03 Female
#> 6 26.01 Female
# 2. Run quantile_plot plot function
quantile_plot(
data = weight_sex,
my_shape = "fill_circle",
point_size = 1.5,
conf_int = TRUE,
conf_level = 0.95,
split_panel = "Split_Panel",
legend_pos = "right",
legend_dir = "vertical",
sci_fill_color = "Sci_NPG",
sci_color_alpha = 0.75,
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::quantile_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/quantile_plot.html.
# Get help with command in R console.
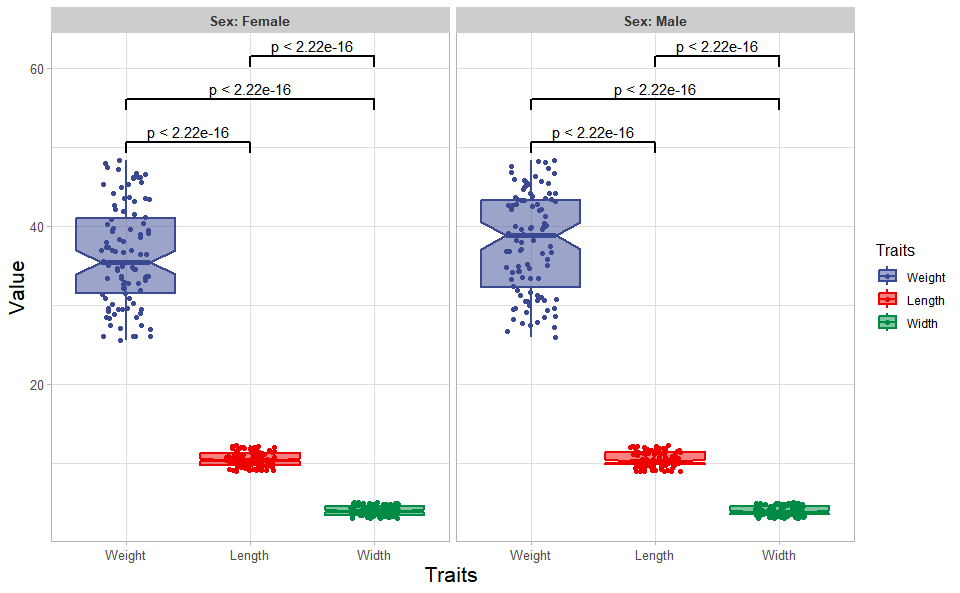
# ?TOmicsVis::quantile_plotInput Data: Dataframe: Length, Width, Weight, and Sex traits dataframe (1st-col: Value, 2nd-col: Traits, 3rd-col: Sex).
Output Plot: Plot: Box plot support two levels and multiple groups with P value.
# 1. Load example datasets
data(traits_sex)
head(traits_sex)
#> Value Traits Sex
#> 1 36.74 Weight Female
#> 2 38.54 Weight Female
#> 3 44.91 Weight Female
#> 4 43.53 Weight Female
#> 5 39.03 Weight Female
#> 6 26.01 Weight Female
# 2. Run box_plot plot function
box_plot(
data = traits_sex,
test_method = "t.test",
test_label = "p.format",
notch = TRUE,
group_level = "Three_Column",
add_element = "jitter",
my_shape = "fill_circle",
sci_fill_color = "Sci_AAAS",
sci_fill_alpha = 0.5,
sci_color_alpha = 1,
legend_pos = "right",
legend_dir = "vertical",
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::box_plot or reference
page https://benben-miao.github.io/TOmicsVis/reference/box_plot.html.
# Get help with command in R console.
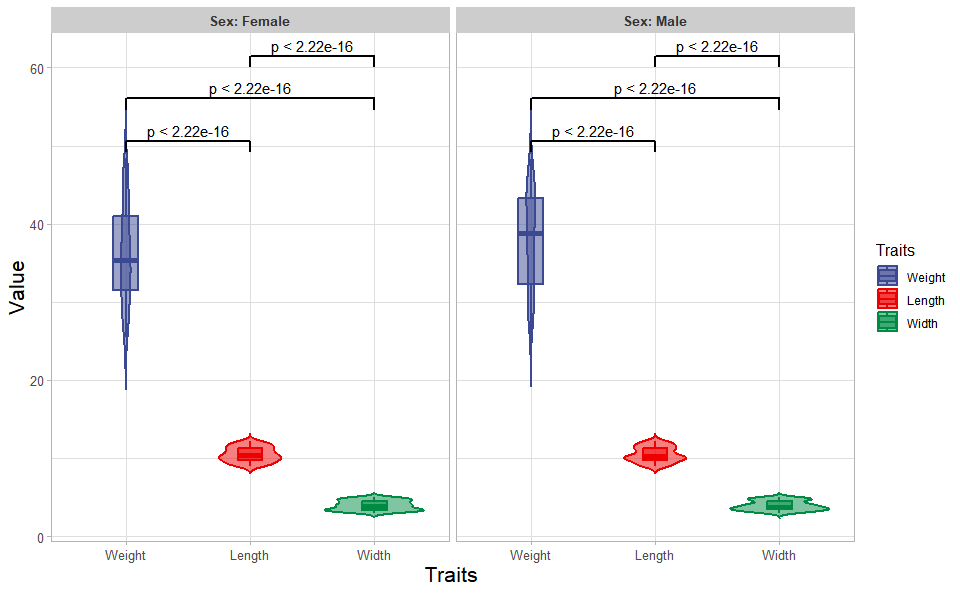
# ?TOmicsVis::box_plotInput Data: Dataframe: Length, Width, Weight, and Sex traits dataframe (1st-col: Value, 2nd-col: Traits, 3rd-col: Sex).
Output Plot: Plot: Violin plot support two levels and multiple groups with P value.
# 1. Load example datasets
data(traits_sex)
# 2. Run violin_plot plot function
violin_plot(
data = traits_sex,
test_method = "t.test",
test_label = "p.format",
group_level = "Three_Column",
violin_orientation = "vertical",
add_element = "boxplot",
element_alpha = 0.5,
my_shape = "plus_times",
sci_fill_color = "Sci_AAAS",
sci_fill_alpha = 0.5,
sci_color_alpha = 1,
legend_pos = "right",
legend_dir = "vertical",
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::violin_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/violin_plot.html.
# Get help with command in R console.
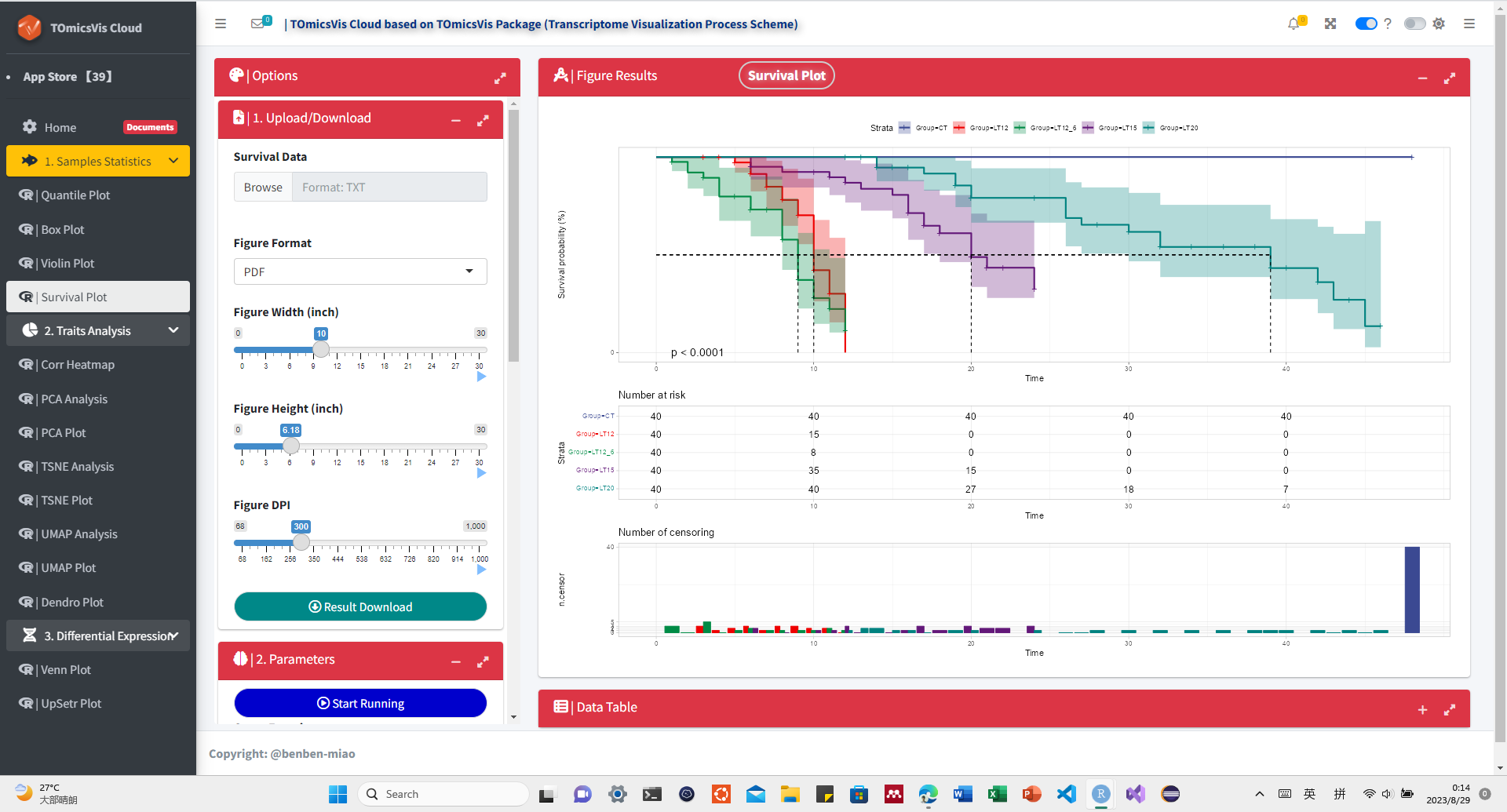
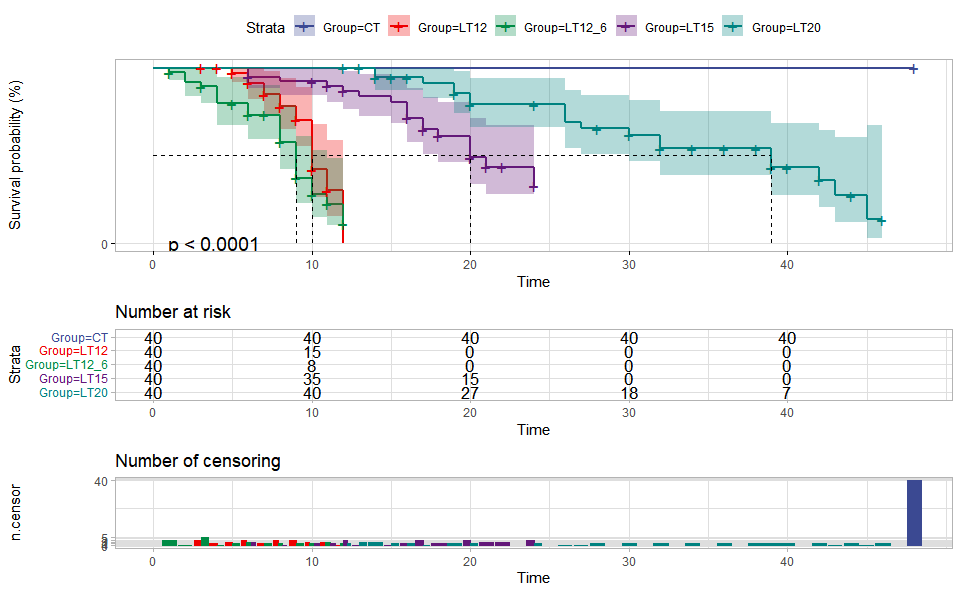
# ?TOmicsVis::violin_plotInput Data: Dataframe: survival record data (1st-col: Time, 2nd-col: Status, 3rd-col: Group).
Output Plot: Survival plot for analyzing and visualizing survival data.
# 1. Load example datasets
data(survival_data)
head(survival_data)
#> Time Status Group
#> 1 48 0 CT
#> 2 48 0 CT
#> 3 48 0 CT
#> 4 48 0 CT
#> 5 48 0 CT
#> 6 48 0 CT
# 2. Run survival_plot plot function
survival_plot(
data = survival_data,
curve_function = "pct",
conf_inter = TRUE,
interval_style = "ribbon",
risk_table = TRUE,
num_censor = TRUE,
sci_palette = "aaas",
ggTheme = "theme_light",
x_start = 0,
y_start = 0,
y_end = 100,
x_break = 10,
y_break = 10
)
Get help using command ?TOmicsVis::survival_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/survival_plot.html.
# Get help with command in R console.
# ?TOmicsVis::survival_plotInput Data: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Output Plot: Plot: heatmap plot filled with Pearson correlation values and P values.
# 1. Load example dataset
data(gene_expression)
head(gene_expression)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2
#> 1 transcript_0 655.78 631.08 669.89 654.21 402.56 447.09 510.08 442.22
#> 2 transcript_1 92.72 112.26 150.30 88.35 76.35 94.55 120.24 80.89
#> 3 transcript_10 21.74 31.11 22.58 15.09 13.67 13.24 12.48 7.53
#> 4 transcript_100 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 transcript_1000 0.00 14.15 36.01 0.00 0.00 193.59 208.45 0.00
#> 6 transcript_10000 89.18 158.04 86.28 82.97 117.78 102.24 129.61 112.73
#> LT15_3 LT12_1 LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 399.82 483.30 437.89 444.06 405.43 416.63 464.75
#> 2 73.94 96.25 82.62 85.48 65.12 61.94 73.44
#> 3 13.35 11.16 11.36 6.96 7.82 4.01 10.02
#> 4 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 232.40 148.58 0.00 181.61 0.02 12.18 0.00
#> 6 85.70 80.89 124.11 115.25 113.87 107.69 119.83
# 2. Run corr_heatmap plot function
corr_heatmap(
data = gene_expression,
corr_method = "pearson",
cell_shape = "square",
fill_type = "full",
lable_size = 3,
axis_angle = 45,
axis_size = 12,
lable_digits = 3,
color_low = "blue",
color_mid = "white",
color_high = "red",
outline_color = "white",
ggTheme = "theme_light"
)
#> Scale for fill is already present.
#> Adding another scale for fill, which will replace the existing scale.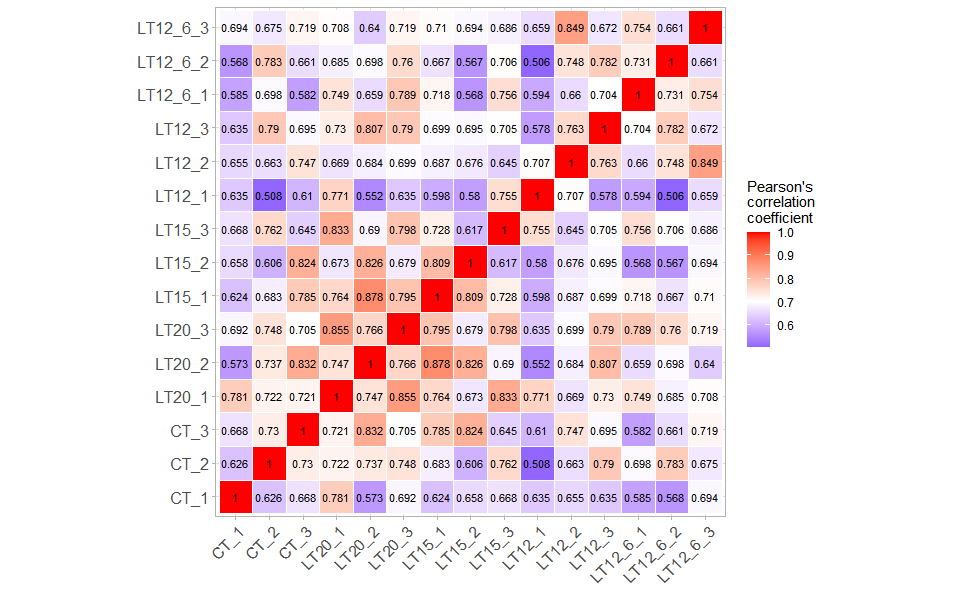
Get help using command ?TOmicsVis::corr_heatmap or
reference page https://benben-miao.github.io/TOmicsVis/reference/corr_heatmap.html.
# Get help with command in R console.
# ?TOmicsVis::corr_heatmapInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Table: PCA dimensional reduction analysis for RNA-Seq.
# 1. Load example datasets
data(gene_expression)
data(samples_groups)
head(samples_groups)
#> Samples Groups
#> 1 CT_1 CT
#> 2 CT_2 CT
#> 3 CT_3 CT
#> 4 LT20_1 LT20
#> 5 LT20_2 LT20
#> 6 LT20_3 LT20
# 2. Run pca_analysis plot function
res <- pca_analysis(gene_expression, samples_groups)
head(res)
#> PC1 PC2 PC3 PC4 PC5 PC6
#> CT_1 -27010.536 -18328.2803 5955.2569 46547.7319 11394.1043 -7197.285
#> CT_2 16248.651 29132.9251 -824.1857 20747.9618 -18798.8755 21096.088
#> CT_3 22421.017 -26832.3964 6789.4490 5864.1171 -15375.3418 17424.861
#> LT20_1 -18587.073 -472.9036 -21638.7836 7765.9575 114.1225 -3943.968
#> LT20_2 33275.933 -9874.9959 -14991.3942 -7443.9250 -4600.8302 -8072.298
#> LT20_3 -1596.255 11683.5426 -10892.8493 381.0795 11080.3560 -8994.187
#> PC7 PC8 PC9 PC10 PC11 PC12
#> CT_1 2150.6739 4850.320 4051.745 7666.9445 -3141.9327 -2487.939
#> CT_2 -12329.1138 -3353.734 4805.659 1503.8533 11184.0296 -4865.436
#> CT_3 12744.2255 -10037.516 -11468.842 202.4016 -11001.6260 -3847.291
#> LT20_1 8864.7482 -14171.127 -1968.082 -3562.1899 7446.2105 14831.486
#> LT20_2 -941.3943 -5072.401 5345.106 6494.1383 -3954.2153 9351.346
#> LT20_3 7263.9321 -7774.725 -1853.546 -21427.2641 -46.1503 -12507.011
#> PC13 PC14 PC15
#> CT_1 -2704.613 2396.7383 2.528517e-11
#> CT_2 -2633.057 -1375.3352 6.825657e-11
#> CT_3 5193.978 188.5601 2.255671e-11
#> LT20_1 3937.457 -7871.8062 4.864246e-11
#> LT20_2 -12904.673 6071.6618 -2.020696e-10
#> LT20_3 -5369.380 2606.1762 1.903509e-11Get help using command ?TOmicsVis::pca_analysis or
reference page https://benben-miao.github.io/TOmicsVis/reference/pca_analysis.html.
# Get help with command in R console.
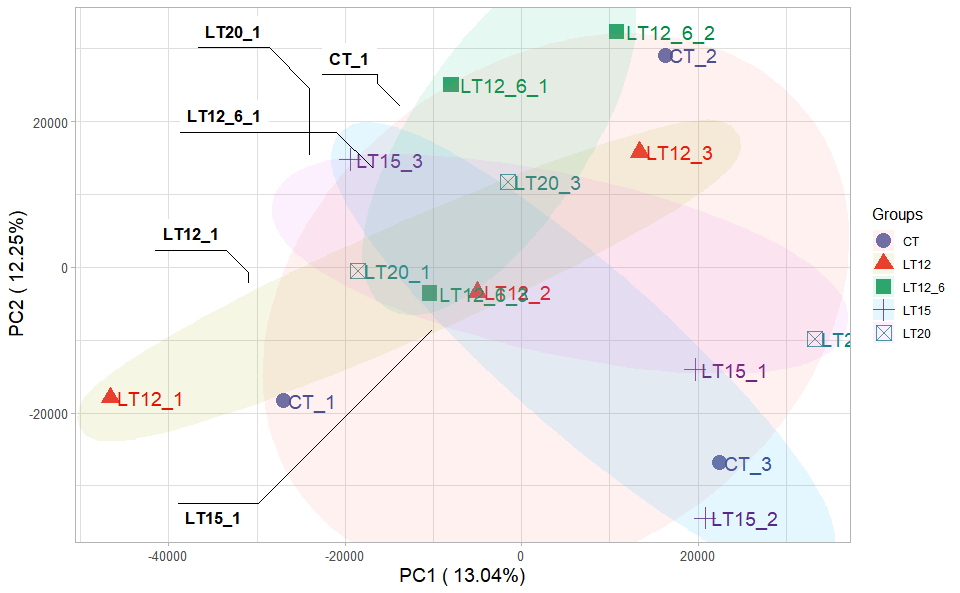
# ?TOmicsVis::pca_analysisInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Plot: Plot: PCA dimensional reduction visualization for RNA-Seq.
# 1. Load example datasets
data(gene_expression)
data(samples_groups)
head(samples_groups)
#> Samples Groups
#> 1 CT_1 CT
#> 2 CT_2 CT
#> 3 CT_3 CT
#> 4 LT20_1 LT20
#> 5 LT20_2 LT20
#> 6 LT20_3 LT20
# 2. Run pca_plot plot function
pca_plot(
sample_gene = gene_expression,
group_sample = samples_groups,
xPC = 1,
yPC = 2,
point_size = 5,
text_size = 5,
fill_alpha = 0.10,
border_alpha = 0.00,
legend_pos = "right",
legend_dir = "vertical",
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::pca_plot or reference
page https://benben-miao.github.io/TOmicsVis/reference/pca_plot.html.
# Get help with command in R console.
# ?TOmicsVis::pca_plotInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Table: TSNE analysis for analyzing and visualizing TSNE algorithm.
# 1. Load example datasets
data(gene_expression)
data(samples_groups)
# 2. Run tsne_analysis plot function
res <- tsne_analysis(gene_expression, samples_groups)
head(res)
#> TSNE1 TSNE2
#> 1 -67.41252 -16.61397
#> 2 43.08349 -34.02654
#> 3 123.32273 54.14358
#> 4 -42.52065 -31.30027
#> 5 94.98790 48.97986
#> 6 -23.90637 -22.26434Get help using command ?TOmicsVis::tsne_analysis or
reference page https://benben-miao.github.io/TOmicsVis/reference/tsne_analysis.html.
# Get help with command in R console.
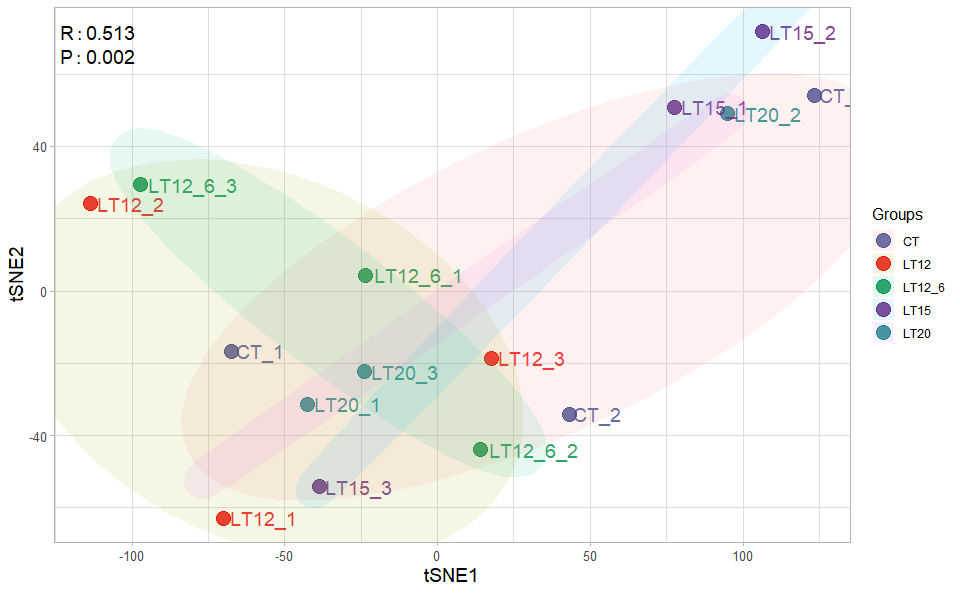
# ?TOmicsVis::tsne_analysisInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Plot: TSNE plot for analyzing and visualizing TSNE algorithm.
# 1. Load example datasets
data(gene_expression)
data(samples_groups)
# 2. Run tsne_plot plot function
tsne_plot(
sample_gene = gene_expression,
group_sample = samples_groups,
seed = 1,
multi_shape = FALSE,
point_size = 5,
point_alpha = 0.8,
text_size = 5,
text_alpha = 0.80,
fill_alpha = 0.10,
border_alpha = 0.00,
sci_fill_color = "Sci_AAAS",
legend_pos = "right",
legend_dir = "vertical",
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::tsne_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/tsne_plot.html.
# Get help with command in R console.
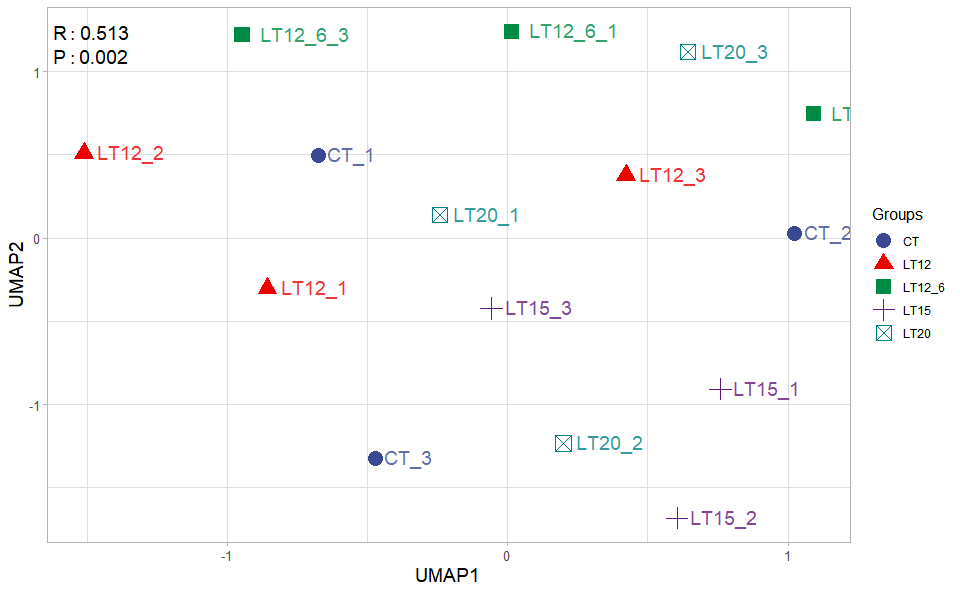
# ?TOmicsVis::tsne_plotInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Table: UMAP analysis for analyzing RNA-Seq data.
# 1. Load example datasets
data(gene_expression)
data(samples_groups)
# 2. Run tsne_plot plot function
res <- umap_analysis(gene_expression, samples_groups)
head(res)
#> UMAP1 UMAP2
#> CT_1 -0.6752746 0.49425898
#> CT_2 1.0232441 0.03062202
#> CT_3 -0.4722297 -1.32183550
#> LT20_1 -0.2414214 0.13870703
#> LT20_2 0.1991701 -1.23434000
#> LT20_3 0.6431577 1.11879669Get help using command ?TOmicsVis::umap_analysis or
reference page https://benben-miao.github.io/TOmicsVis/reference/umap_analysis.html.
# Get help with command in R console.
# ?TOmicsVis::umap_analysisInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Plot: UMAP plot for analyzing and visualizing UMAP algorithm.
# 1. Load example datasets
data(gene_expression)
data(samples_groups)
# 2. Run tsne_plot plot function
umap_plot(
sample_gene = gene_expression,
group_sample = samples_groups,
seed = 1,
multi_shape = TRUE,
point_size = 5,
point_alpha = 1,
text_size = 5,
text_alpha = 0.80,
fill_alpha = 0.00,
border_alpha = 0.00,
sci_fill_color = "Sci_AAAS",
legend_pos = "right",
legend_dir = "vertical",
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::umap_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/umap_plot.html.
# Get help with command in R console.
# ?TOmicsVis::umap_plotInput Data: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Output Plot: Plot: dendrogram for multiple samples clustering.
# 1. Load example datasets
data(gene_expression)
# 2. Run plot function
dendro_plot(
data = gene_expression,
dist_method = "euclidean",
hc_method = "ward.D2",
tree_type = "rectangle",
k_num = 5,
palette = "npg",
color_labels_by_k = TRUE,
horiz = FALSE,
label_size = 1,
line_width = 1,
rect = TRUE,
rect_fill = TRUE,
xlab = "Samples",
ylab = "Height",
ggTheme = "theme_light"
)
#> Registered S3 method overwritten by 'dendextend':
#> method from
#> rev.hclust vegan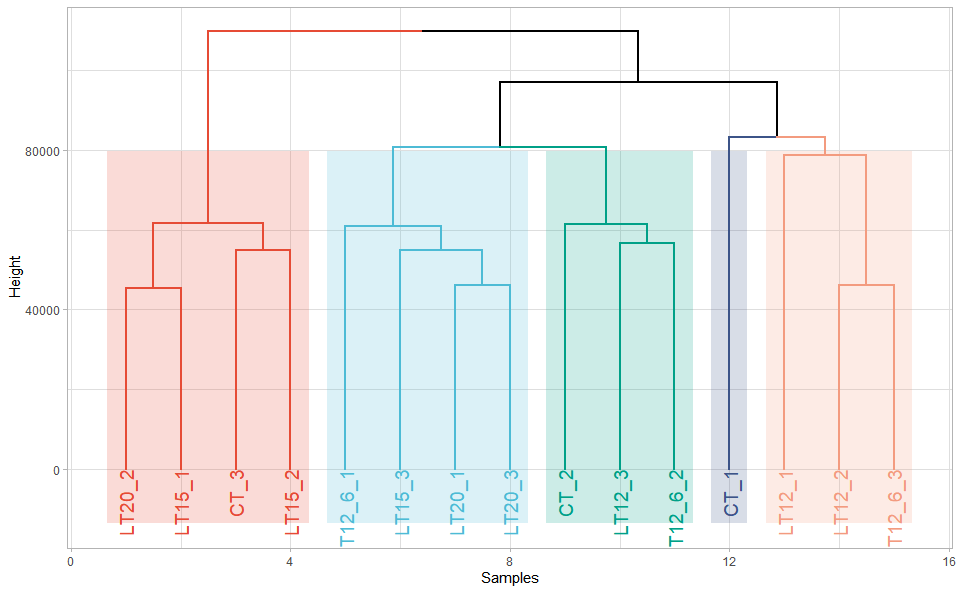
Get help using command ?TOmicsVis::dendro_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/dendro_plot.html.
# Get help with command in R console.
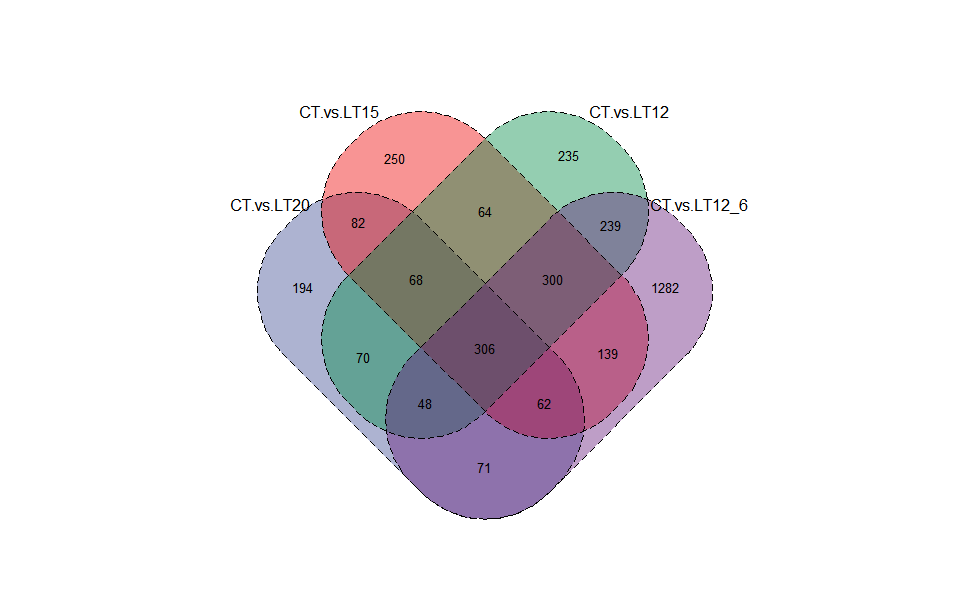
# ?TOmicsVis::dendro_plotInput Data2: Dataframe: Paired comparisons differentially expressed genes (degs) among groups (1st-col~: degs of paired comparisons).
Output Plot: Venn plot for stat common and unique gene among multiple sets.
# 1. Load example datasets
data(degs_lists)
head(degs_lists)
#> CT.vs.LT20 CT.vs.LT15 CT.vs.LT12 CT.vs.LT12_6
#> 1 transcript_9024 transcript_4738 transcript_9956 transcript_10354
#> 2 transcript_604 transcript_6050 transcript_7601 transcript_2959
#> 3 transcript_3912 transcript_1039 transcript_5960 transcript_5919
#> 4 transcript_8676 transcript_1344 transcript_3240 transcript_2395
#> 5 transcript_8832 transcript_3069 transcript_10224 transcript_9881
#> 6 transcript_74 transcript_9809 transcript_3151 transcript_8836
# 2. Run venn_plot plot function
venn_plot(
data = degs_lists,
title_size = 1,
label_show = TRUE,
label_size = 0.8,
border_show = TRUE,
line_type = "longdash",
ellipse_shape = "circle",
sci_fill_color = "Sci_AAAS",
sci_fill_alpha = 0.65
)
Get help using command ?TOmicsVis::venn_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/venn_plot.html.
# Get help with command in R console.
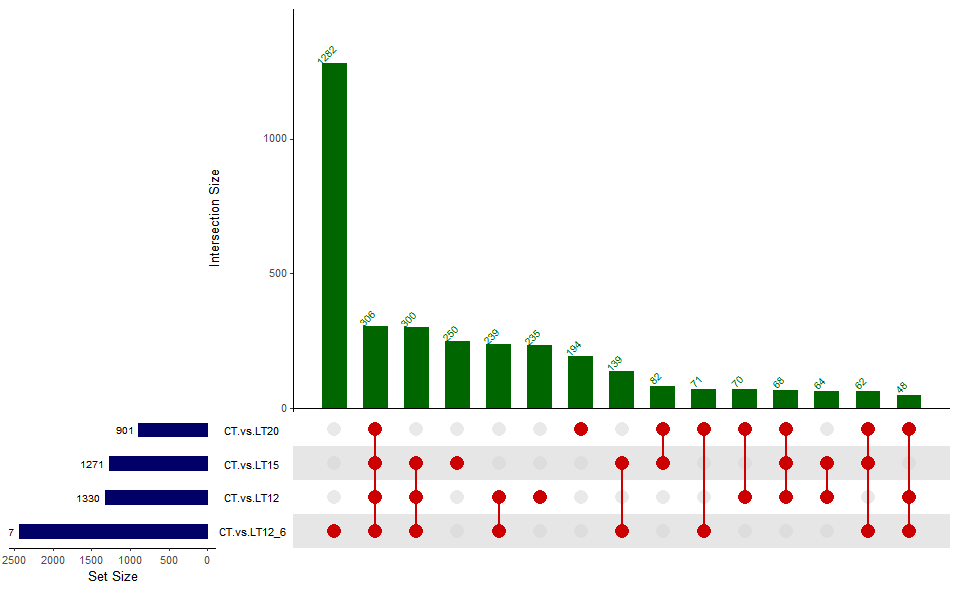
# ?TOmicsVis::venn_plotInput Data2: Dataframe: Paired comparisons differentially expressed genes (degs) among groups (1st-col~: degs of paired comparisons).
Output Plot: UpSet plot for stat common and unique gene among multiple sets.
# 1. Load example datasets
data(degs_lists)
head(degs_lists)
#> CT.vs.LT20 CT.vs.LT15 CT.vs.LT12 CT.vs.LT12_6
#> 1 transcript_9024 transcript_4738 transcript_9956 transcript_10354
#> 2 transcript_604 transcript_6050 transcript_7601 transcript_2959
#> 3 transcript_3912 transcript_1039 transcript_5960 transcript_5919
#> 4 transcript_8676 transcript_1344 transcript_3240 transcript_2395
#> 5 transcript_8832 transcript_3069 transcript_10224 transcript_9881
#> 6 transcript_74 transcript_9809 transcript_3151 transcript_8836
# 2. Run upsetr_plot plot function
upsetr_plot(
data = degs_lists,
sets_num = 4,
keep_order = FALSE,
order_by = "freq",
decrease = TRUE,
mainbar_color = "#006600",
number_angle = 45,
matrix_color = "#cc0000",
point_size = 4.5,
point_alpha = 0.5,
line_size = 0.8,
shade_color = "#cdcdcd",
shade_alpha = 0.5,
setsbar_color = "#000066",
setsnum_size = 6,
text_scale = 1.2
)
Get help using command ?TOmicsVis::upsetr_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/upsetr_plot.html.
# Get help with command in R console.
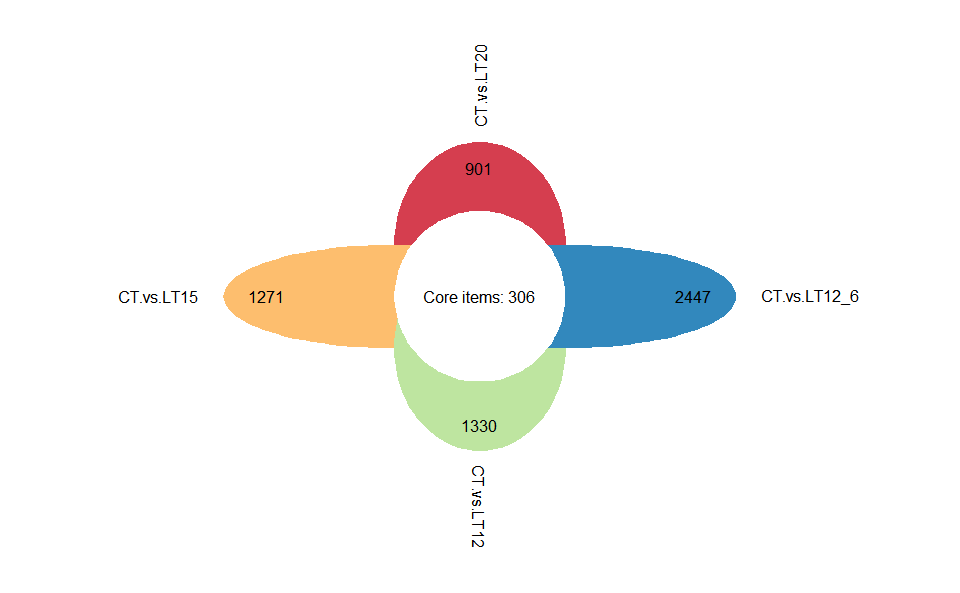
# ?TOmicsVis::upsetr_plotInput Data2: Dataframe: Paired comparisons differentially expressed genes (degs) among groups (1st-col~: degs of paired comparisons).
Output Plot: Flower plot for stat common and unique gene among multiple sets.
# 1. Load example datasets
data(degs_lists)
# 2. Run plot function
flower_plot(
flower_dat = degs_lists,
angle = 90,
a = 1,
b = 2,
r = 1,
ellipse_col_pal = "Spectral",
circle_col = "white",
label_text_cex = 1
)
Get help using command ?TOmicsVis::flower_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/flower_plot.html.
# Get help with command in R console.
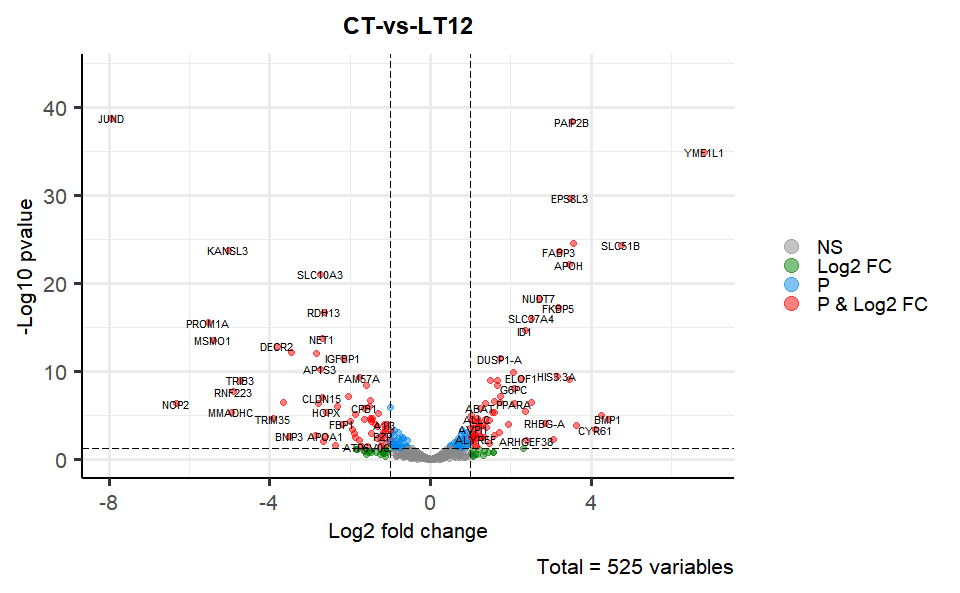
# ?TOmicsVis::flower_plotInput Data2: Dataframe: All DEGs of paired comparison CT-vs-LT12 stats dataframe (1st-col: Genes, 2nd-col: log2FoldChange, 3rd-col: Pvalue, 4th-col: FDR).
Output Plot: Volcano plot for visualizing differentailly expressed genes.
# 1. Load example datasets
data(degs_stats)
head(degs_stats)
#> Gene log2FoldChange Pvalue FDR
#> 1 A1I3 -1.13855748 0.000111040 0.000862478
#> 2 A1M 0.59076131 0.070988041 0.192551708
#> 3 A2M 0.09297827 0.819706797 0.913893947
#> 4 A2ML1 -0.26940689 0.745374782 0.874295125
#> 5 ABAT 1.24811621 0.000001440 0.000016800
#> 6 ABCC3 -0.72947545 0.005171574 0.024228298
# 2. Run volcano_plot plot function
volcano_plot(
data = degs_stats,
title = "CT-vs-LT12",
log2fc_cutoff = 1,
pq_value = "pvalue",
pq_cutoff = 0.05,
cutoff_line = "longdash",
point_shape = "large_circle",
point_size = 2,
point_alpha = 0.5,
color_normal = "#888888",
color_log2fc = "#008000",
color_pvalue = "#0088ee",
color_Log2fc_p = "#ff0000",
label_size = 3,
boxed_labels = FALSE,
draw_connectors = FALSE,
legend_pos = "right"
)
Get help using command ?TOmicsVis::volcano_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/volcano_plot.html.
# Get help with command in R console.
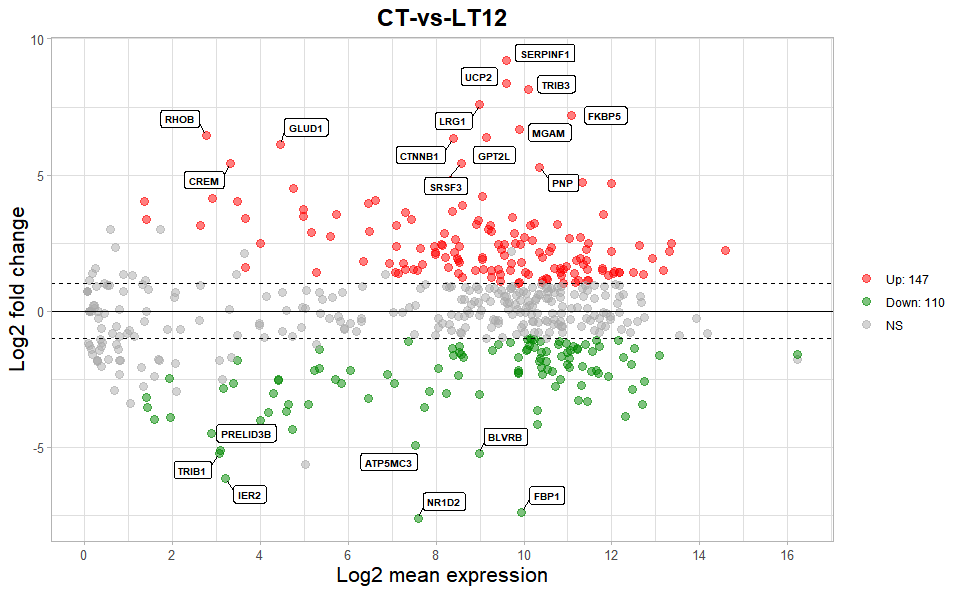
# ?TOmicsVis::volcano_plotInput Data2: Dataframe: All DEGs of paired comparison CT-vs-LT12 stats2 dataframe (1st-col: Gene, 2nd-col: baseMean, 3rd-col: Log2FoldChange, 4th-col: FDR).
Output Plot: MversusA plot for visualizing differentially expressed genes.
# 1. Load example datasets
data(degs_stats2)
head(degs_stats2)
#> name baseMean log2FoldChange padj
#> 1 A1I3 0.1184475 0.0000000 NA
#> 2 A1M 1654.4618140 0.6789538 5.280802e-02
#> 3 A2M 681.0463277 1.5263838 3.920000e-07
#> 4 A2ML1 389.7226640 3.8933573 1.180000e-14
#> 5 ABAT 364.7810090 -2.3554014 1.559230e-04
#> 6 ABCC3 1.1346239 1.2932740 4.491812e-01
# 2. Run volcano_plot plot function
ma_plot(
data = degs_stats2,
foldchange = 2,
fdr_value = 0.05,
point_size = 3.0,
color_up = "#FF0000",
color_down = "#008800",
color_alpha = 0.5,
top_method = "fc",
top_num = 20,
label_size = 8,
label_box = TRUE,
title = "CT-vs-LT12",
xlab = "Log2 mean expression",
ylab = "Log2 fold change",
ggTheme = "theme_light"
)
Get help using command ?TOmicsVis::ma_plot or reference
page https://benben-miao.github.io/TOmicsVis/reference/ma_plot.html.
# Get help with command in R console.
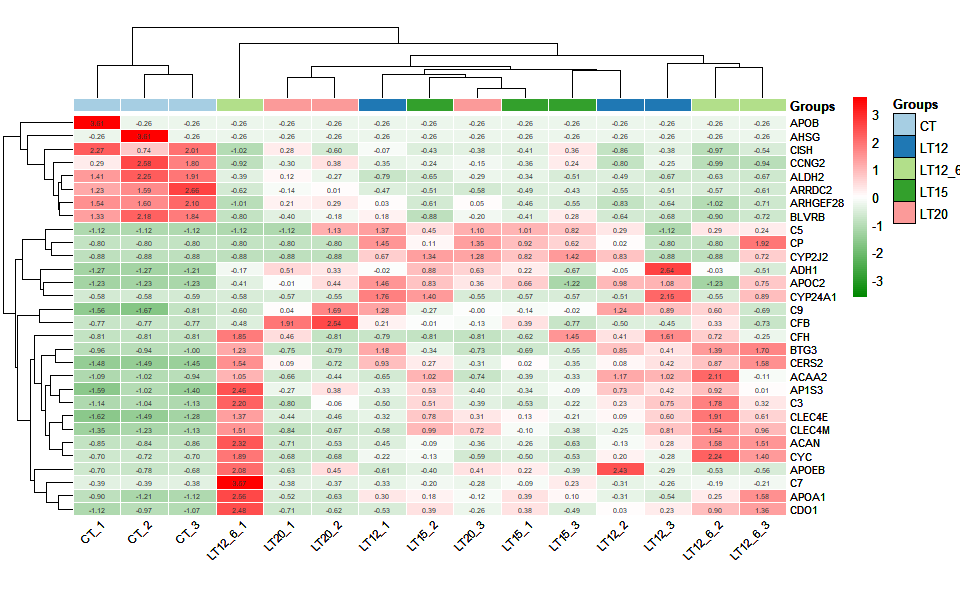
# ?TOmicsVis::ma_plotInput Data1: Dataframe: Shared DEGs of all paired comparisons in all samples expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Plot: Heatmap group for visualizing grouped gene expression data.
# 1. Load example datasets
data(gene_expression2)
data(samples_groups)
# 2. Run heatmap_group plot function
heatmap_group(
sample_gene = gene_expression2[1:30,],
group_sample = samples_groups,
scale_data = "row",
clust_method = "complete",
border_show = TRUE,
border_color = "#ffffff",
value_show = TRUE,
value_decimal = 2,
value_size = 5,
axis_size = 8,
cell_height = 10,
low_color = "#00880055",
mid_color = "#ffffff",
high_color = "#ff000055",
na_color = "#ff8800",
x_angle = 45
)
Get help using command ?TOmicsVis::heatmap_group or
reference page https://benben-miao.github.io/TOmicsVis/reference/heatmap_group.html.
# Get help with command in R console.
# ?TOmicsVis::heatmap_groupInput Data2: Dataframe: Shared DEGs of all paired comparisons in all samples expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~: Samples).
Output Plot: Circos heatmap plot for visualizing gene expressing in multiple samples.
# 1. Load example datasets
data(gene_expression2)
head(gene_expression2)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2 LT15_3 LT12_1
#> 1 ACAA2 24.50 39.83 55.38 114.11 159.32 96.88 169.56 464.84 182.66 116.08
#> 2 ACAN 14.97 18.71 10.30 71.23 142.67 213.54 253.15 320.80 104.15 174.02
#> 3 ADH1 1.54 1.56 2.04 14.95 13.60 15.87 12.80 17.74 6.06 10.97
#> 4 AHSG 0.00 1911.99 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 ALDH2 2.07 2.86 2.54 0.85 0.49 0.47 0.42 0.13 0.26 0.00
#> 6 AP1S3 6.62 14.59 9.30 24.90 33.94 23.19 24.00 36.08 27.40 24.06
#> LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 497.29 464.48 471.43 693.62 229.77
#> 2 305.81 469.48 1291.90 991.90 966.77
#> 3 10.71 30.95 9.84 10.91 7.28
#> 4 0.00 0.00 0.00 0.00 0.00
#> 5 0.28 0.11 0.37 0.15 0.11
#> 6 38.74 34.54 62.72 41.36 28.75
# 2. Run circos_heatmap plot function
circos_heatmap(
data = gene_expression2[1:50,],
low_color = "#0000ff",
mid_color = "#ffffff",
high_color = "#ff0000",
gap_size = 25,
cluster_run = TRUE,
cluster_method = "complete",
distance_method = "euclidean",
dend_show = "inside",
dend_height = 0.2,
track_height = 0.3,
rowname_show = "outside",
rowname_size = 0.8
)
#> Note: 15 points are out of plotting region in sector 'group', track
#> '3'.
#> Note: 15 points are out of plotting region in sector 'group', track
#> '3'.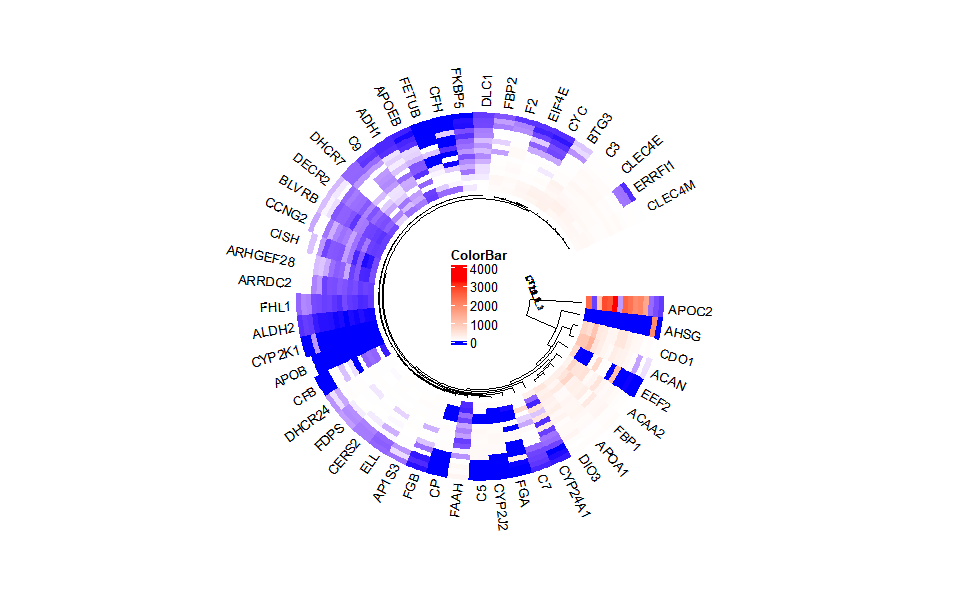
Get help using command ?TOmicsVis::circos_heatmap or
reference page https://benben-miao.github.io/TOmicsVis/reference/circos_heatmap.html.
# Get help with command in R console.
# ?TOmicsVis::circos_heatmapInput Data2: Dataframe: Shared DEGs of all paired comparisons in all samples expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~: Samples).
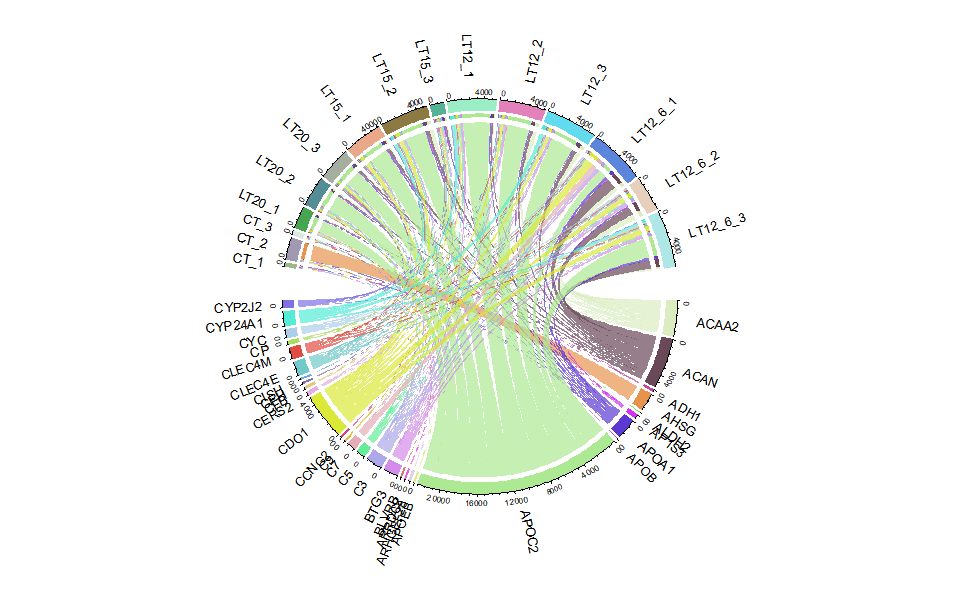
Output Plot: Chord plot for visualizing the relationships of pathways and genes.
# 1. Load chord_data example datasets
data(gene_expression2)
head(gene_expression2)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2 LT15_3 LT12_1
#> 1 ACAA2 24.50 39.83 55.38 114.11 159.32 96.88 169.56 464.84 182.66 116.08
#> 2 ACAN 14.97 18.71 10.30 71.23 142.67 213.54 253.15 320.80 104.15 174.02
#> 3 ADH1 1.54 1.56 2.04 14.95 13.60 15.87 12.80 17.74 6.06 10.97
#> 4 AHSG 0.00 1911.99 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 ALDH2 2.07 2.86 2.54 0.85 0.49 0.47 0.42 0.13 0.26 0.00
#> 6 AP1S3 6.62 14.59 9.30 24.90 33.94 23.19 24.00 36.08 27.40 24.06
#> LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 497.29 464.48 471.43 693.62 229.77
#> 2 305.81 469.48 1291.90 991.90 966.77
#> 3 10.71 30.95 9.84 10.91 7.28
#> 4 0.00 0.00 0.00 0.00 0.00
#> 5 0.28 0.11 0.37 0.15 0.11
#> 6 38.74 34.54 62.72 41.36 28.75
# 2. Run chord_plot plot function
chord_plot(
data = gene_expression2[1:30,],
multi_colors = "VividColors",
color_seed = 10,
color_alpha = 0.3,
link_visible = TRUE,
link_dir = -1,
link_type = "diffHeight",
sector_scale = "Origin",
width_circle = 3,
dist_name = 3,
label_dir = "Vertical",
dist_label = 0.3,
label_scale = 0.8
)
#> rn cn value1 value2 o1 o2 x1 x2 col
#> 1 ACAA2 CT_1 24.50 24.50 15 30 3779.75 394.66 #DAECC0B2
#> 2 ACAN CT_1 14.97 14.97 15 29 5349.40 370.16 #694858B2
#> 3 ADH1 CT_1 1.54 1.54 15 28 166.82 355.19 #C047A3B2
#> 4 AHSG CT_1 0.00 0.00 15 27 1911.99 353.65 #E7934EB2
#> 5 ALDH2 CT_1 2.07 2.07 15 26 11.11 353.65 #71EC4AB2
#> 6 AP1S3 CT_1 6.62 6.62 15 25 430.19 351.58 #D334EDB2Get help using command ?TOmicsVis::chord_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/chord_plot.html.
# Get help with command in R console.
# ?TOmicsVis::chord_plotInput Data: Dataframe: All DEGs of paired comparison CT-vs-LT12 stats dataframe (1st-col: Genes, 2nd-col: log2FoldChange, 3rd-col: Pvalue, 4th-col: FDR).
Output Plot: Gene cluster trend plot for visualizing gene expression trend profile in multiple samples.
# 1. Load example datasets
data(degs_stats)
# 2. Run plot function
gene_rank_plot(
data = degs_stats,
log2fc = 1,
palette = "Spectral",
top_n = 10,
genes_to_label = NULL,
label_size = 5,
base_size = 12,
title = "Gene ranking dotplot",
xlab = "Ranking of differentially expressed genes",
ylab = "Log2FoldChange"
)![]()
Get help using command ?TOmicsVis::gene_rank_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/gene_rank_plot.html.
# Get help with command in R console.
# ?TOmicsVis::gene_rank_plotInput Data2: Dataframe: Shared DEGs of all paired comparisons in all groups expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~n-1-col: Groups, n-col: Pathways).
Output Plot: Gene cluster trend plot for visualizing gene expression trend profile in multiple samples.
# 1. Load example datasets
data(gene_expression3)
# 2. Run plot function
gene_cluster_trend(
data = gene_expression3[,-7],
thres = 0.25,
min_std = 0.2,
palette = "PiYG",
cluster_num = 4
)
#> 0 genes excluded.
#> 0 genes excluded.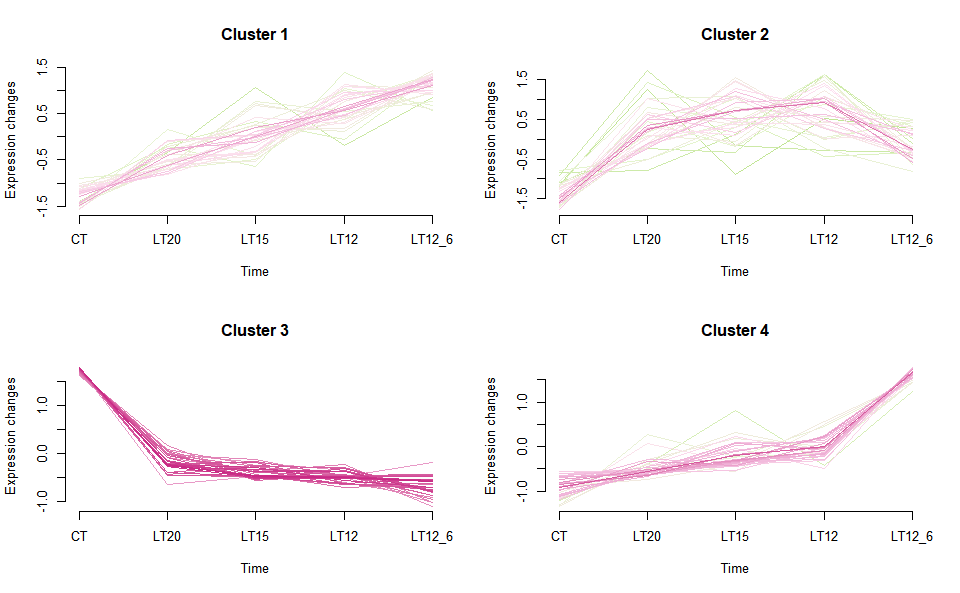
#> NULLGet help using command ?TOmicsVis::gene_cluster_trend or
reference page https://benben-miao.github.io/TOmicsVis/reference/gene_cluster_trend.html.
# Get help with command in R console.
# ?TOmicsVis::gene_cluster_trendInput Data2: Dataframe: Shared DEGs of all paired comparisons in all groups expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~n-1-col: Groups, n-col: Pathways).
Output Plot: Trend plot for visualizing gene expression trend profile in multiple traits.
# 1. Load example datasets
data(gene_expression3)
head(gene_expression3)
#> Genes CT LT20 LT15 LT12 LT12_6
#> 1 ACAA2 39.903333 123.4366667 272.3533 359.28333 464.940000
#> 2 ACAN 14.660000 142.4800000 226.0333 316.43667 1083.523333
#> 3 ADH1 1.713333 14.8066667 12.2000 17.54333 9.343333
#> 4 AHSG 637.330000 0.0000000 0.0000 0.00000 0.000000
#> 5 ALDH2 2.490000 0.6033333 0.2700 0.13000 0.210000
#> 6 AP1S3 10.170000 27.3433333 29.1600 32.44667 44.276667
#> Pathways
#> 1 PPAR signaling pathway
#> 2 PPAR signaling pathway
#> 3 PPAR signaling pathway
#> 4 PPAR signaling pathway
#> 5 PPAR signaling pathway
#> 6 PPAR signaling pathway
# 2. Run trend_plot plot function
trend_plot(
data = gene_expression3[1:100,],
scale_method = "centerObs",
miss_value = "exclude",
line_alpha = 0.5,
show_points = TRUE,
show_boxplot = TRUE,
num_column = 1,
xlab = "Traits",
ylab = "Genes Expression",
sci_fill_color = "Sci_AAAS",
sci_fill_alpha = 0.8,
sci_color_alpha = 0.8,
legend_pos = "right",
legend_dir = "vertical",
ggTheme = "theme_light"
)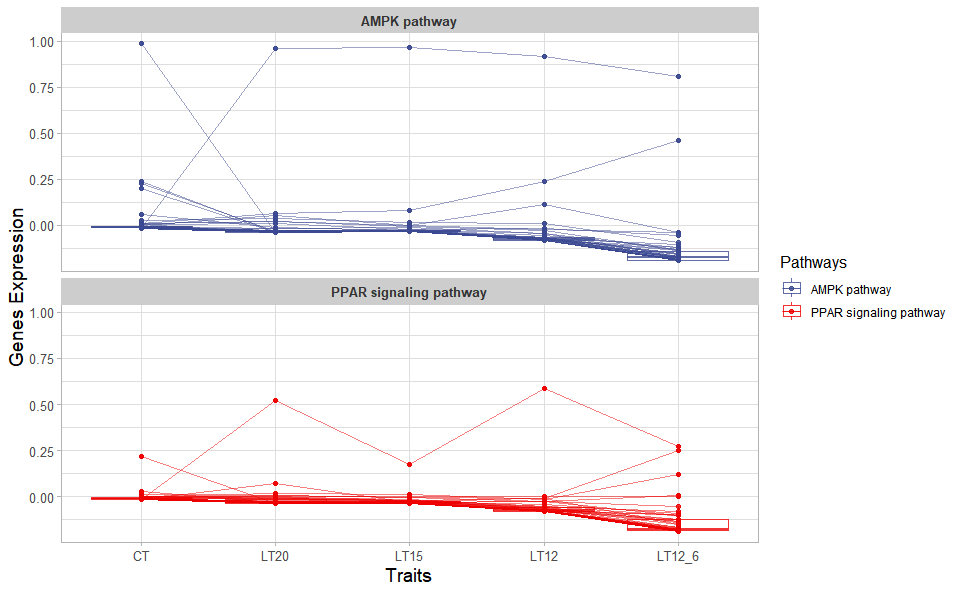
Get help using command ?TOmicsVis::trend_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/trend_plot.html.
# Get help with command in R console.
# ?TOmicsVis::trend_plotInput Data1: Dataframe: All genes in all samples expression dataframe of RNA-Seq (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
Output Plot: WGCNA analysis pipeline for RNA-Seq.
# 1. Load wgcna_pipeline example datasets
data(gene_expression)
head(gene_expression)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2
#> 1 transcript_0 655.78 631.08 669.89 654.21 402.56 447.09 510.08 442.22
#> 2 transcript_1 92.72 112.26 150.30 88.35 76.35 94.55 120.24 80.89
#> 3 transcript_10 21.74 31.11 22.58 15.09 13.67 13.24 12.48 7.53
#> 4 transcript_100 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 transcript_1000 0.00 14.15 36.01 0.00 0.00 193.59 208.45 0.00
#> 6 transcript_10000 89.18 158.04 86.28 82.97 117.78 102.24 129.61 112.73
#> LT15_3 LT12_1 LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 399.82 483.30 437.89 444.06 405.43 416.63 464.75
#> 2 73.94 96.25 82.62 85.48 65.12 61.94 73.44
#> 3 13.35 11.16 11.36 6.96 7.82 4.01 10.02
#> 4 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 232.40 148.58 0.00 181.61 0.02 12.18 0.00
#> 6 85.70 80.89 124.11 115.25 113.87 107.69 119.83
data(samples_groups)
head(samples_groups)
#> Samples Groups
#> 1 CT_1 CT
#> 2 CT_2 CT
#> 3 CT_3 CT
#> 4 LT20_1 LT20
#> 5 LT20_2 LT20
#> 6 LT20_3 LT20
# 2. Run wgcna_pipeline plot function
# wgcna_pipeline(gene_expression[1:3000,], samples_groups)Get help using command ?TOmicsVis::wgcna_pipeline or
reference page https://benben-miao.github.io/TOmicsVis/reference/wgcna_pipeline.html.
# Get help with command in R console.
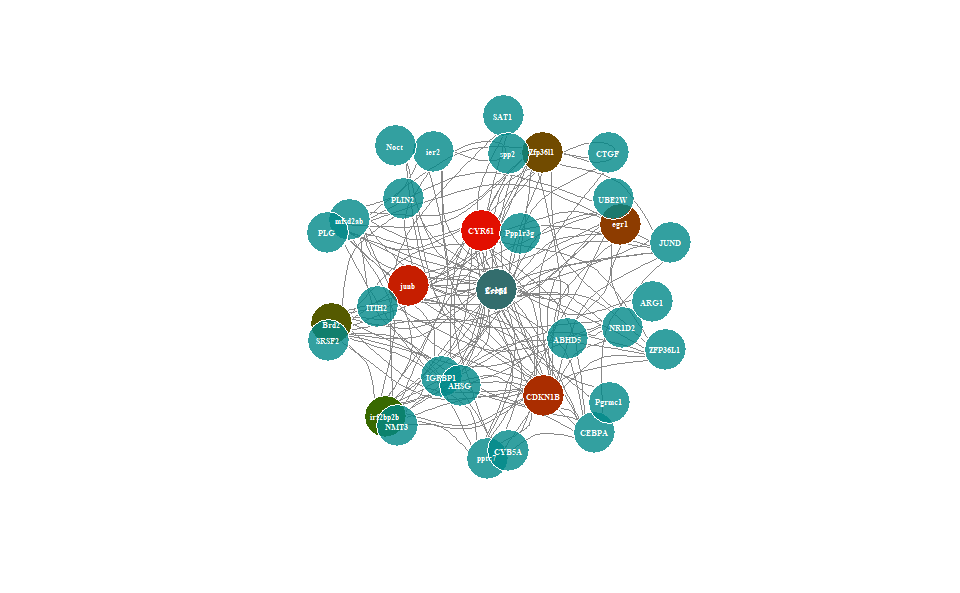
# ?TOmicsVis::wgcna_pipelineInput Data: Dataframe: Network data from WGCNA tan module top-200 dataframe (1st-col: Source, 2nd-col: Target).
Output Plot: Network plot for analyzing and visualizing relationship of genes.
# 1. Load example datasets
data(network_data)
head(network_data)
#> Source Target
#> 1 Cebpd Cebpd
#> 2 CYR61 Cebpd
#> 3 Cebpd CDKN1B
#> 4 CYR61 CDKN1B
#> 5 junb Cebpd
#> 6 IGFBP1 Cebpd
# 2. Run network_plot plot function
network_plot(
data = network_data,
calc_by = "degree",
degree_value = 0.5,
normal_color = "#008888cc",
border_color = "#FFFFFF",
from_color = "#FF0000cc",
to_color = "#008800cc",
normal_shape = "circle",
spatial_shape = "circle",
node_size = 25,
lable_color = "#FFFFFF",
label_size = 0.5,
edge_color = "#888888",
edge_width = 1.5,
edge_curved = TRUE,
net_layout = "layout_on_sphere"
)
Get help using command ?TOmicsVis::network_plot or
reference page https://benben-miao.github.io/TOmicsVis/reference/network_plot.html.
# Get help with command in R console.
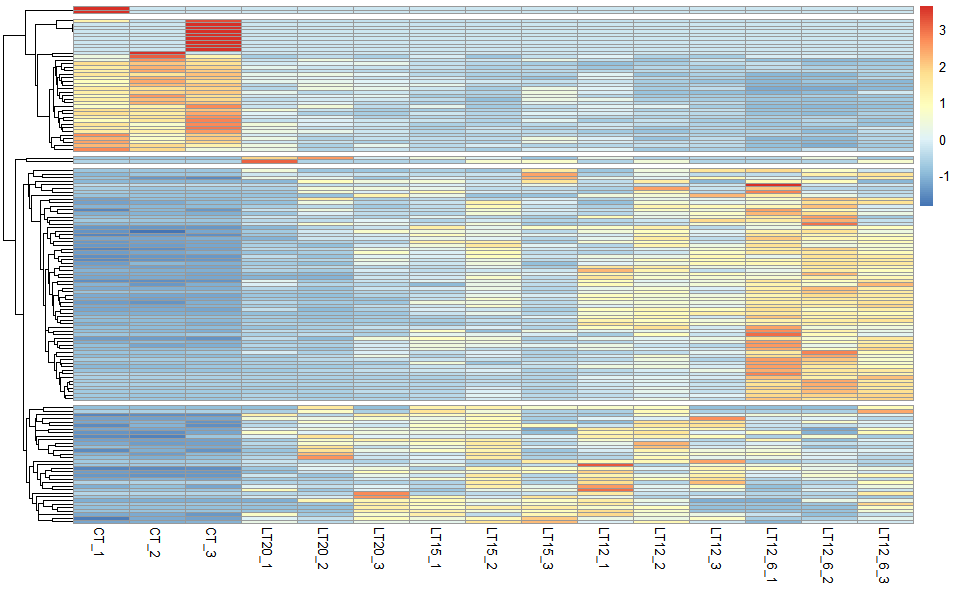
# ?TOmicsVis::network_plotInput Data: Dataframe: Shared DEGs of all paired comparisons in all samples expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~: Samples).
Output Plot: Heatmap cluster plot for visualizing clustered gene expression data.
# 1. Load example datasets
data(gene_expression2)
head(gene_expression2)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2 LT15_3 LT12_1
#> 1 ACAA2 24.50 39.83 55.38 114.11 159.32 96.88 169.56 464.84 182.66 116.08
#> 2 ACAN 14.97 18.71 10.30 71.23 142.67 213.54 253.15 320.80 104.15 174.02
#> 3 ADH1 1.54 1.56 2.04 14.95 13.60 15.87 12.80 17.74 6.06 10.97
#> 4 AHSG 0.00 1911.99 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 ALDH2 2.07 2.86 2.54 0.85 0.49 0.47 0.42 0.13 0.26 0.00
#> 6 AP1S3 6.62 14.59 9.30 24.90 33.94 23.19 24.00 36.08 27.40 24.06
#> LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 497.29 464.48 471.43 693.62 229.77
#> 2 305.81 469.48 1291.90 991.90 966.77
#> 3 10.71 30.95 9.84 10.91 7.28
#> 4 0.00 0.00 0.00 0.00 0.00
#> 5 0.28 0.11 0.37 0.15 0.11
#> 6 38.74 34.54 62.72 41.36 28.75
# 2. Run network_plot plot function
heatmap_cluster(
data = gene_expression2,
dist_method = "euclidean",
hc_method = "average",
k_num = 5,
show_rownames = FALSE,
palette = "RdBu",
cluster_pal = "Set1",
border_color = "#ffffff",
angle_col = 45,
label_size = 10,
base_size = 12,
line_color = "#0000cd",
line_alpha = 0.2,
summary_color = "#0000cd",
summary_alpha = 0.8
)
#> Using Cluster, gene as id variables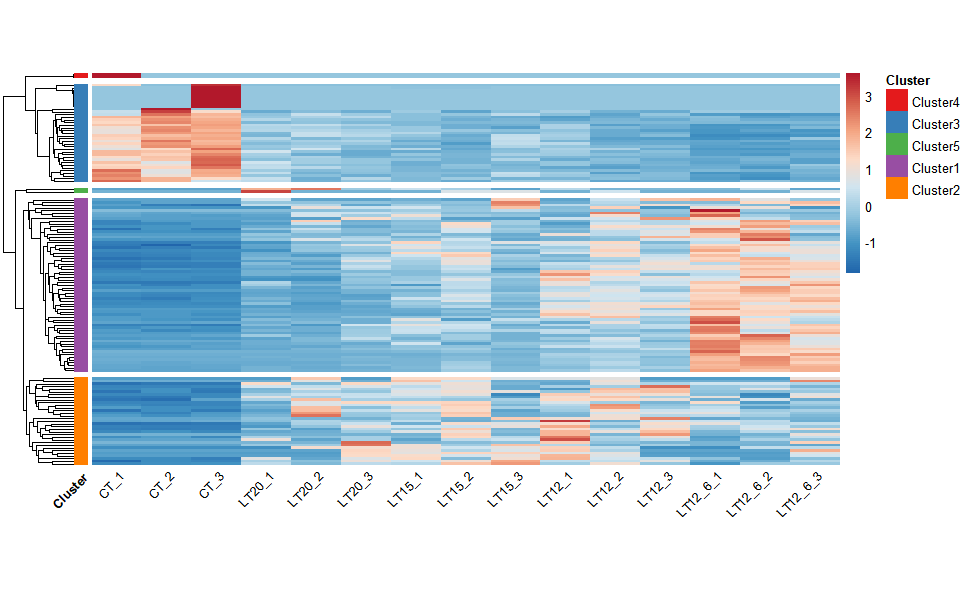
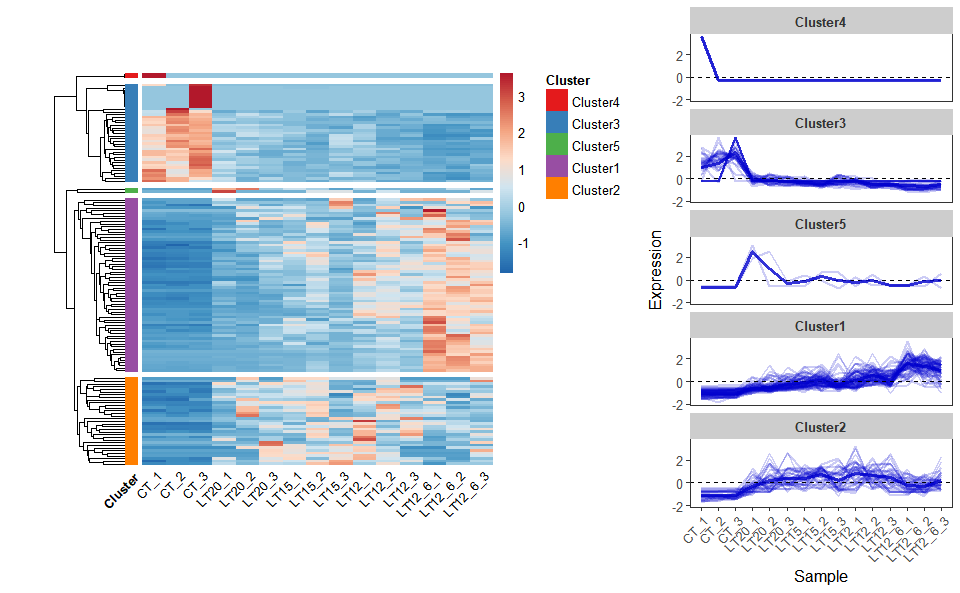
Get help using command ?TOmicsVis::heatmap_cluster or
reference page https://benben-miao.github.io/TOmicsVis/reference/heatmap_cluster.html.
# Get help with command in R console.
# ?TOmicsVis::heatmap_clusterInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Table: GO enrichment analysis based on GO annotation results (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
head(gene_go_kegg)
#> Genes
#> 1 FN1
#> 2 14-3-3ZETA
#> 3 A1I3
#> 4 A2M
#> 5 AARS
#> 6 ABAT
#> biological_process
#> 1 GO:0003181(atrioventricular valve morphogenesis);GO:0003128(heart field specification);GO:0001756(somitogenesis)
#> 2 <NA>
#> 3 <NA>
#> 4 <NA>
#> 5 GO:0006419(alanyl-tRNA aminoacylation)
#> 6 GO:0009448(gamma-aminobutyric acid metabolic process)
#> cellular_component
#> 1 GO:0005576(extracellular region)
#> 2 <NA>
#> 3 GO:0005615(extracellular space)
#> 4 GO:0005615(extracellular space)
#> 5 GO:0005737(cytoplasm)
#> 6 <NA>
#> molecular_function
#> 1 <NA>
#> 2 GO:0019904(protein domain specific binding)
#> 3 GO:0004866(endopeptidase inhibitor activity)
#> 4 GO:0004866(endopeptidase inhibitor activity)
#> 5 GO:0004813(alanine-tRNA ligase activity);GO:0005524(ATP binding);GO:0000049(tRNA binding);GO:0008270(zinc ion binding)
#> 6 GO:0003867(4-aminobutyrate transaminase activity);GO:0030170(pyridoxal phosphate binding)
#> kegg_pathway
#> 1 ko04810(Regulation of actin cytoskeleton);ko04510(Focal adhesion);ko04151(PI3K-Akt signaling pathway);ko04512(ECM-receptor interaction)
#> 2 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko04610(Complement and coagulation cascades)
#> 5 ko00970(Aminoacyl-tRNA biosynthesis)
#> 6 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
# 2. Run go_enrich analysis function
res <- go_enrich(
go_anno = gene_go_kegg[,-5],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05
)
head(res)
#> ID ontology
#> 1 GO:0000221 cellular component
#> 2 GO:0000275 cellular component
#> 3 GO:0000276 cellular component
#> 4 GO:0000398 biological process
#> 5 GO:0000774 molecular function
#> 6 GO:0001671 molecular function
#> Description
#> 1 vacuolar proton-transporting V-type ATPase, V1 domain
#> 2 mitochondrial proton-transporting ATP synthase complex, catalytic core F
#> 3 mitochondrial proton-transporting ATP synthase complex, coupling factor F
#> 4 mRNA splicing, via spliceosome
#> 5 adenyl-nucleotide exchange factor activity
#> 6 ATPase activator activity
#> GeneRatio BgRatio pvalue p.adjust qvalue
#> 1 1/101 1/1279 7.896794e-02 1.110997e-01 9.458955e-02
#> 2 1/101 1/1279 7.896794e-02 1.110997e-01 9.458955e-02
#> 3 6/101 6/1279 2.109128e-07 1.075656e-05 9.158058e-06
#> 4 1/101 14/1279 6.858207e-01 7.363549e-01 6.269275e-01
#> 5 1/101 1/1279 7.896794e-02 1.110997e-01 9.458955e-02
#> 6 1/101 1/1279 7.896794e-02 1.110997e-01 9.458955e-02
#> geneID Count
#> 1 ATP6V1H 1
#> 2 ATP5F1E 1
#> 3 ATP5MC1/ATP5ME/ATP5MG/ATP5PB/ATP5PD/ATP5PF 6
#> 4 CDC40 1
#> 5 BAG2 1
#> 6 ATP1B1 1Get help using command ?TOmicsVis::go_enrich or
reference page https://benben-miao.github.io/TOmicsVis/reference/go_enrich.html.
# Get help with command in R console.
# ?TOmicsVis::go_enrichInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: GO enrichment analysis and stat plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run go_enrich_stat analysis function
go_enrich_stat(
go_anno = gene_go_kegg[,-5],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
max_go_item = 15,
strip_fill = "#CDCDCD",
xtext_angle = 45,
sci_fill_color = "Sci_AAAS",
sci_fill_alpha = 0.8,
ggTheme = "theme_light"
)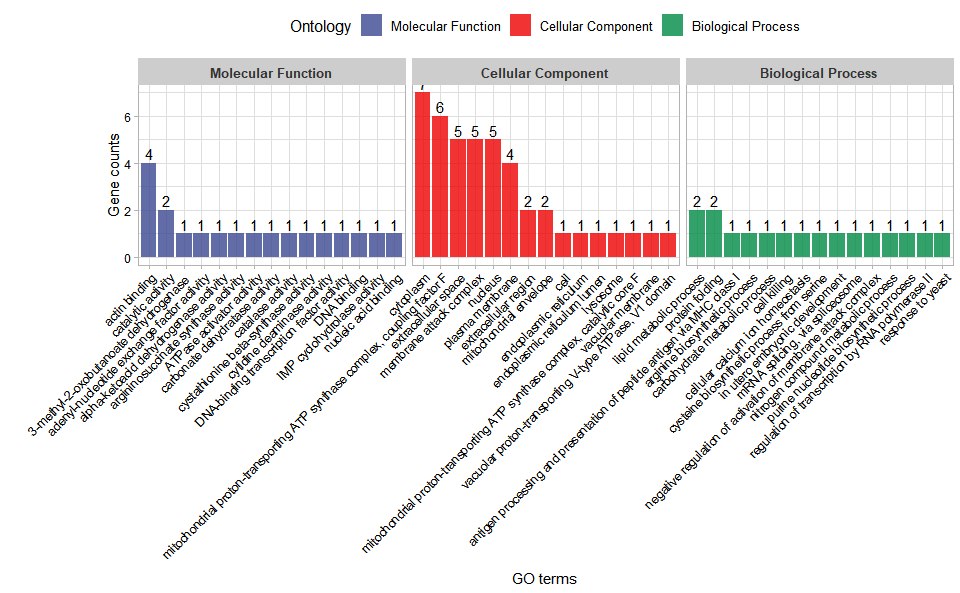
Get help using command ?TOmicsVis::go_enrich_stat or
reference page https://benben-miao.github.io/TOmicsVis/reference/go_enrich_stat.html.
# Get help with command in R console.
# ?TOmicsVis::go_enrich_statInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: GO enrichment analysis and bar plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run go_enrich_bar analysis function
go_enrich_bar(
go_anno = gene_go_kegg[,-5],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
sign_by = "p.adjust",
category_num = 30,
font_size = 12,
low_color = "#ff0000aa",
high_color = "#008800aa",
ggTheme = "theme_light"
)
#> Scale for fill is already present.
#> Adding another scale for fill, which will replace the existing scale.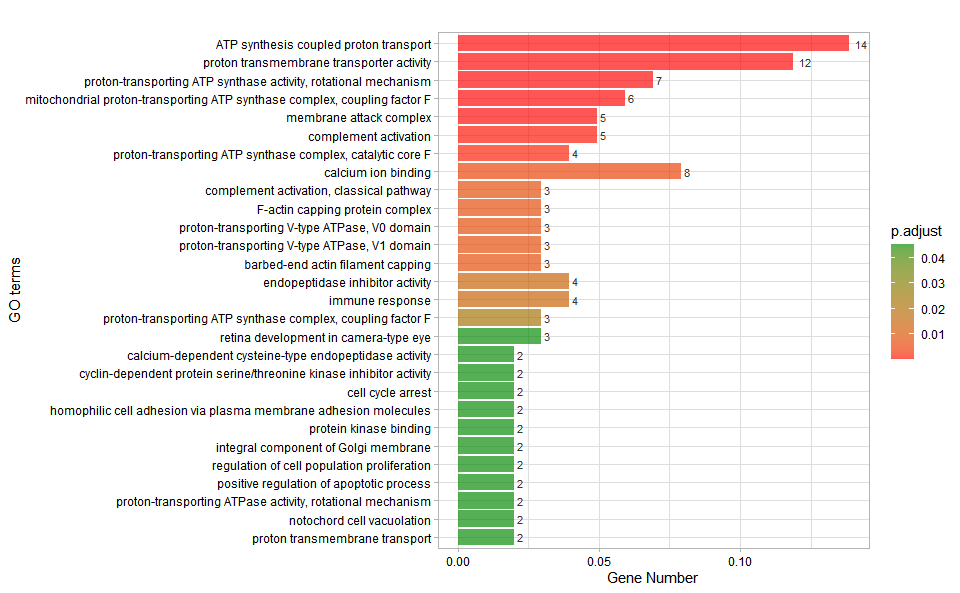
Get help using command ?TOmicsVis::go_enrich_bar or
reference page https://benben-miao.github.io/TOmicsVis/reference/go_enrich_bar.html.
# Get help with command in R console.
# ?TOmicsVis::go_enrich_barInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: GO enrichment analysis and dot plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run go_enrich_dot analysis function
go_enrich_dot(
go_anno = gene_go_kegg[,-5],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
sign_by = "p.adjust",
category_num = 30,
font_size = 12,
low_color = "#ff0000aa",
high_color = "#008800aa",
ggTheme = "theme_light"
)
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.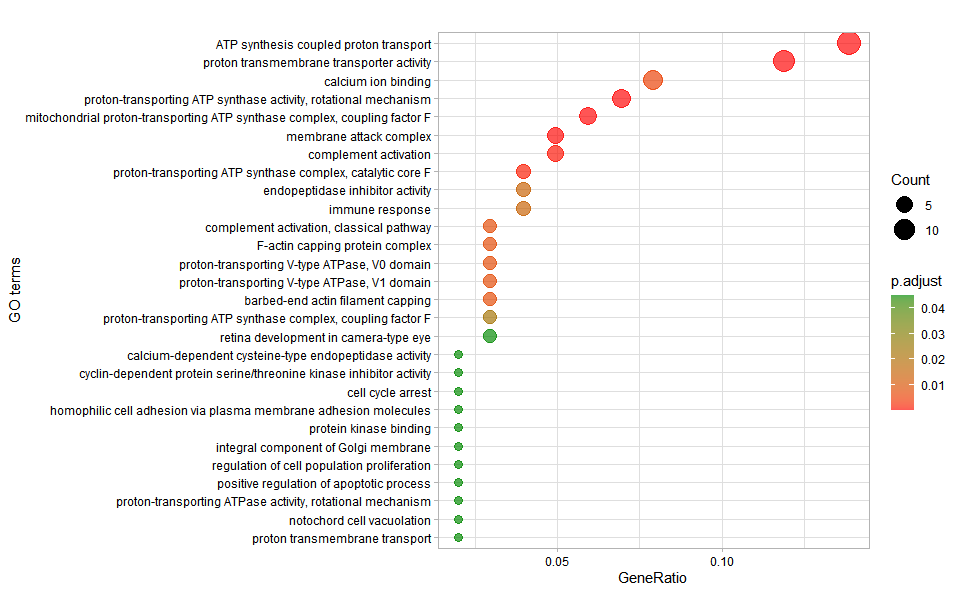
Get help using command ?TOmicsVis::go_enrich_dot or
reference page https://benben-miao.github.io/TOmicsVis/reference/go_enrich_dot.html.
# Get help with command in R console.
# ?TOmicsVis::go_enrich_dotInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: GO enrichment analysis and net plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run go_enrich_net analysis function
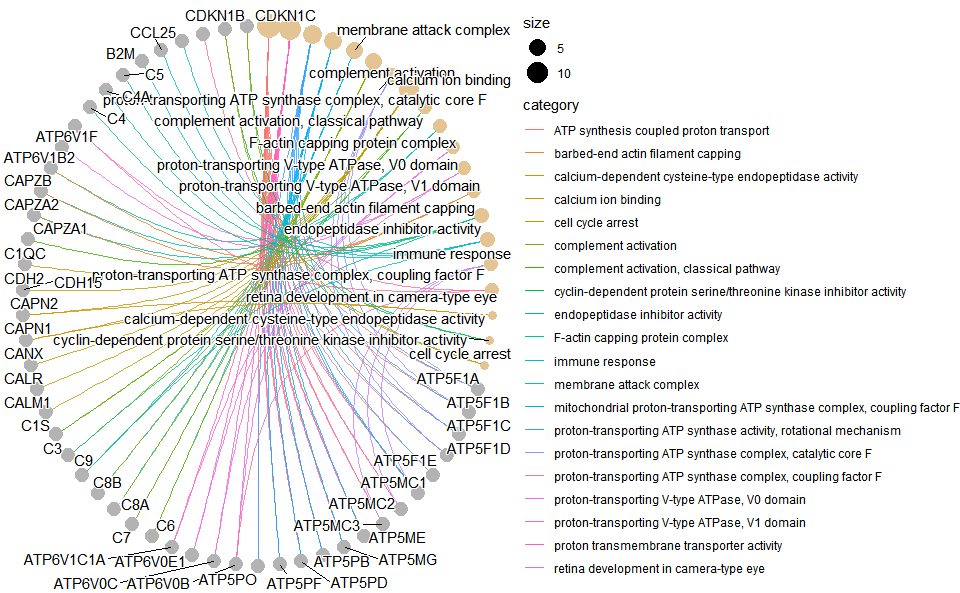
go_enrich_net(
go_anno = gene_go_kegg[,-5],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
category_num = 20,
net_layout = "circle",
net_circular = TRUE,
low_color = "#ff0000aa",
high_color = "#008800aa"
)
Get help using command ?TOmicsVis::go_enrich_net or
reference page https://benben-miao.github.io/TOmicsVis/reference/go_enrich_net.html.
# Get help with command in R console.
# ?TOmicsVis::go_enrich_netInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: GO enrichment analysis based on GO annotation results (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
head(gene_go_kegg)
#> Genes
#> 1 FN1
#> 2 14-3-3ZETA
#> 3 A1I3
#> 4 A2M
#> 5 AARS
#> 6 ABAT
#> biological_process
#> 1 GO:0003181(atrioventricular valve morphogenesis);GO:0003128(heart field specification);GO:0001756(somitogenesis)
#> 2 <NA>
#> 3 <NA>
#> 4 <NA>
#> 5 GO:0006419(alanyl-tRNA aminoacylation)
#> 6 GO:0009448(gamma-aminobutyric acid metabolic process)
#> cellular_component
#> 1 GO:0005576(extracellular region)
#> 2 <NA>
#> 3 GO:0005615(extracellular space)
#> 4 GO:0005615(extracellular space)
#> 5 GO:0005737(cytoplasm)
#> 6 <NA>
#> molecular_function
#> 1 <NA>
#> 2 GO:0019904(protein domain specific binding)
#> 3 GO:0004866(endopeptidase inhibitor activity)
#> 4 GO:0004866(endopeptidase inhibitor activity)
#> 5 GO:0004813(alanine-tRNA ligase activity);GO:0005524(ATP binding);GO:0000049(tRNA binding);GO:0008270(zinc ion binding)
#> 6 GO:0003867(4-aminobutyrate transaminase activity);GO:0030170(pyridoxal phosphate binding)
#> kegg_pathway
#> 1 ko04810(Regulation of actin cytoskeleton);ko04510(Focal adhesion);ko04151(PI3K-Akt signaling pathway);ko04512(ECM-receptor interaction)
#> 2 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko04610(Complement and coagulation cascades)
#> 5 ko00970(Aminoacyl-tRNA biosynthesis)
#> 6 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
# 2. Run go_enrich analysis function
res <- kegg_enrich(
kegg_anno = gene_go_kegg[,c(1,5)],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05
)
head(res)
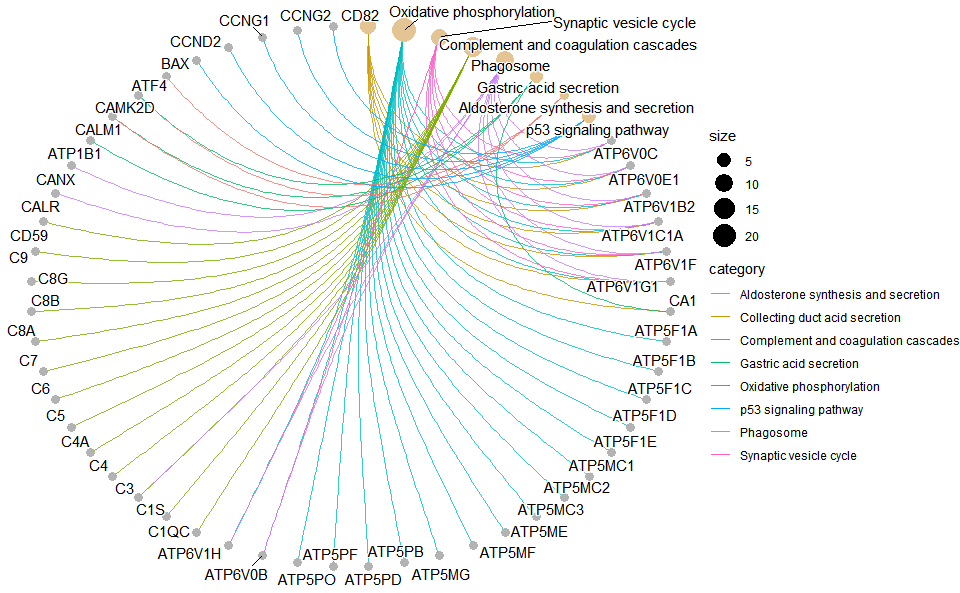
#> ID Description GeneRatio BgRatio
#> ko04966 ko04966 Collecting duct acid secretion 7/101 7/1279
#> ko00190 ko00190 Oxidative phosphorylation 23/101 88/1279
#> ko04721 ko04721 Synaptic vesicle cycle 8/101 13/1279
#> ko04610 ko04610 Complement and coagulation cascades 13/101 43/1279
#> ko04145 ko04145 Phagosome 11/101 33/1279
#> ko04971 ko04971 Gastric acid secretion 4/101 4/1279
#> pvalue p.adjust qvalue
#> ko04966 1.573976e-08 2.030430e-06 1.723090e-06
#> ko00190 5.232645e-08 3.375056e-06 2.864185e-06
#> ko04721 1.069634e-06 4.599427e-05 3.903227e-05
#> ko04610 1.078094e-05 3.476853e-04 2.950573e-04
#> ko04145 1.941460e-05 5.008968e-04 4.250776e-04
#> ko04971 3.679084e-05 7.910030e-04 6.712714e-04
#> geneID
#> ko04966 ATP6V0C/ATP6V0E1/ATP6V1B2/ATP6V1C1A/ATP6V1F/ATP6V1G1/CA1
#> ko00190 ATP5F1A/ATP5F1B/ATP5F1C/ATP5F1D/ATP5F1E/ATP5MC1/ATP5MC2/ATP5MC3/ATP5ME/ATP5MF/ATP5MG/ATP5PB/ATP5PD/ATP5PF/ATP5PO/ATP6V0B/ATP6V0C/ATP6V0E1/ATP6V1B2/ATP6V1C1A/ATP6V1F/ATP6V1G1/ATP6V1H
#> ko04721 ATP6V0B/ATP6V0C/ATP6V0E1/ATP6V1B2/ATP6V1C1A/ATP6V1F/ATP6V1G1/ATP6V1H
#> ko04610 C1QC/C1S/C3/C4/C4A/C5/C6/C7/C8A/C8B/C8G/C9/CD59
#> ko04145 ATP6V0B/ATP6V0C/ATP6V0E1/ATP6V1B2/ATP6V1C1A/ATP6V1F/ATP6V1G1/ATP6V1H/C3/CALR/CANX
#> ko04971 ATP1B1/CA1/CALM1/CAMK2D
#> Count
#> ko04966 7
#> ko00190 23
#> ko04721 8
#> ko04610 13
#> ko04145 11
#> ko04971 4Get help using command ?TOmicsVis::kegg_enrich or
reference page https://benben-miao.github.io/TOmicsVis/reference/kegg_enrich.html.
# Get help with command in R console.
# ?TOmicsVis::kegg_enrichInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: KEGG enrichment analysis and bar plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run kegg_enrich_bar analysis function
kegg_enrich_bar(
kegg_anno = gene_go_kegg[,c(1,5)],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
sign_by = "p.adjust",
category_num = 30,
font_size = 12,
low_color = "#ff0000aa",
high_color = "#008800aa",
ggTheme = "theme_light"
)
#> Scale for fill is already present.
#> Adding another scale for fill, which will replace the existing scale.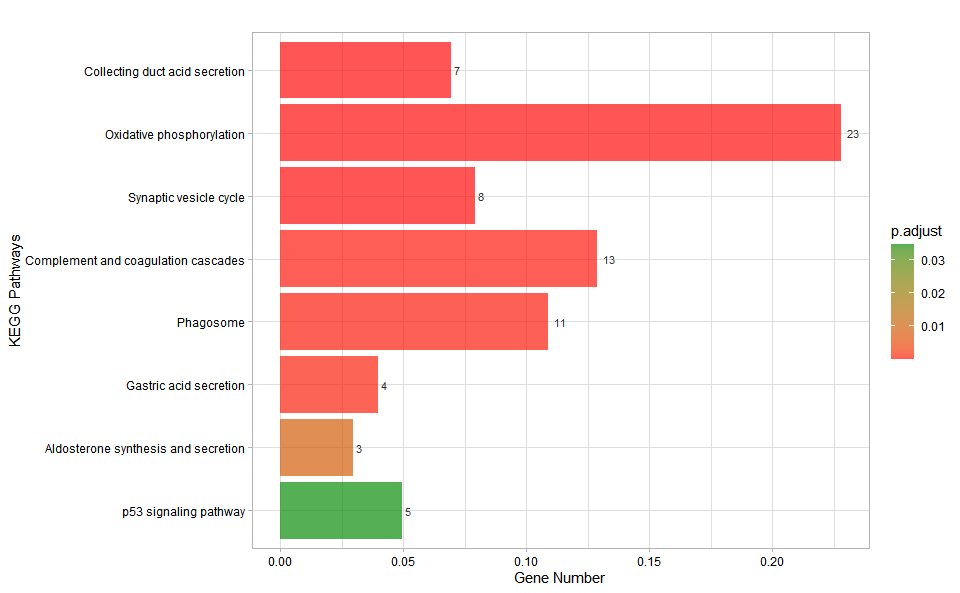
Get help using command ?TOmicsVis::kegg_enrich_bar or
reference page https://benben-miao.github.io/TOmicsVis/reference/kegg_enrich_bar.html.
# Get help with command in R console.
# ?TOmicsVis::kegg_enrich_barInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: KEGG enrichment analysis and dot plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run kegg_enrich_dot analysis function
kegg_enrich_dot(
kegg_anno = gene_go_kegg[,c(1,5)],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
sign_by = "p.adjust",
category_num = 30,
font_size = 12,
low_color = "#ff0000aa",
high_color = "#008800aa",
ggTheme = "theme_light"
)
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.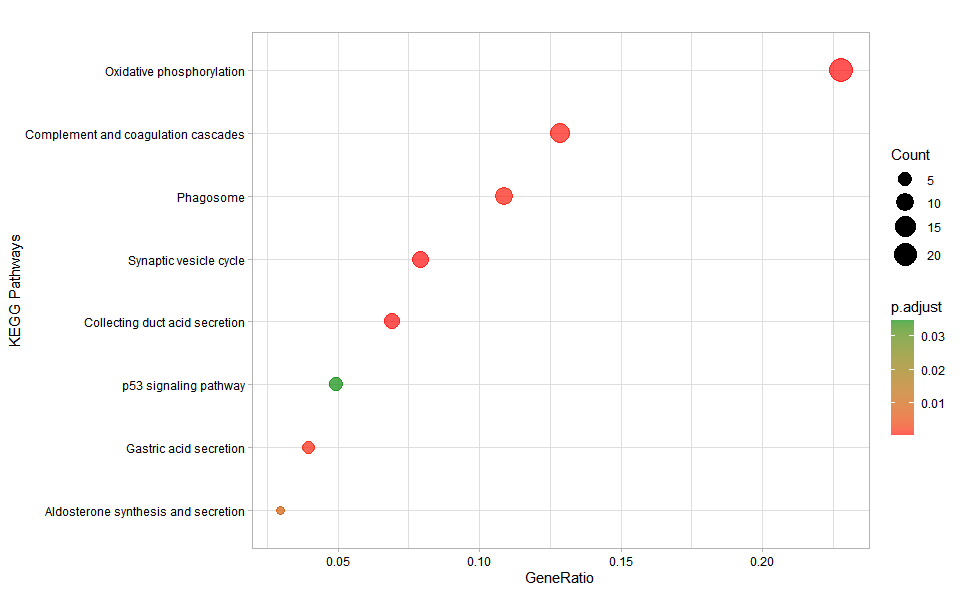
Get help using command ?TOmicsVis::kegg_enrich_dot or
reference page https://benben-miao.github.io/TOmicsVis/reference/kegg_enrich_dot.html.
# Get help with command in R console.
# ?TOmicsVis::kegg_enrich_dotInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: KEGG enrichment analysis and net plot (None/Exist Reference Genome).
# 1. Load example datasets
data(gene_go_kegg)
# 2. Run kegg_enrich_net analysis function
kegg_enrich_net(
kegg_anno = gene_go_kegg[,c(1,5)],
degs_list = gene_go_kegg[100:200,1],
padjust_method = "fdr",
pvalue_cutoff = 0.05,
qvalue_cutoff = 0.05,
category_num = 20,
net_layout = "circle",
net_circular = TRUE,
low_color = "#ff0000aa",
high_color = "#008800aa"
)
Get help using command ?TOmicsVis::kegg_enrich_net or
reference page https://benben-miao.github.io/TOmicsVis/reference/kegg_enrich_net.html.
# Get help with command in R console.
# ?TOmicsVis::kegg_enrich_netInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Table: Table split used for splitting a grouped column to multiple columns.
# 1. Load example datasets
data(gene_go_kegg2)
head(gene_go_kegg2)
#> Genes
#> 1 FN1
#> 2 14-3-3ZETA
#> 3 A1I3
#> 4 A2M
#> 5 AARS
#> 6 ABAT
#> kegg_pathway
#> 1 ko04810(Regulation of actin cytoskeleton);ko04510(Focal adhesion);ko04151(PI3K-Akt signaling pathway);ko04512(ECM-receptor interaction)
#> 2 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko04610(Complement and coagulation cascades)
#> 5 ko00970(Aminoacyl-tRNA biosynthesis)
#> 6 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
#> go_category
#> 1 biological_process
#> 2 biological_process
#> 3 biological_process
#> 4 biological_process
#> 5 biological_process
#> 6 biological_process
#> go_term
#> 1 GO:0003181(atrioventricular valve morphogenesis);GO:0003128(heart field specification);GO:0001756(somitogenesis)
#> 2 <NA>
#> 3 <NA>
#> 4 <NA>
#> 5 GO:0006419(alanyl-tRNA aminoacylation)
#> 6 GO:0009448(gamma-aminobutyric acid metabolic process)
# 2. Run table_split function
res <- table_split(
data = gene_go_kegg2,
grouped_var = "go_category",
value_var = "go_term",
miss_drop = TRUE
)
head(res)
#> Genes
#> 1 14-3-3ZETA
#> 2 A1I3
#> 3 A2M
#> 4 AARS
#> 5 ABAT
#> 6 ABCB7
#> kegg_pathway
#> 1 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 2 ko04610(Complement and coagulation cascades)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko00970(Aminoacyl-tRNA biosynthesis)
#> 5 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
#> 6 ko02010(ABC transporters)
#> biological_process
#> 1 <NA>
#> 2 <NA>
#> 3 <NA>
#> 4 GO:0006419(alanyl-tRNA aminoacylation)
#> 5 GO:0009448(gamma-aminobutyric acid metabolic process)
#> 6 <NA>
#> cellular_component
#> 1 <NA>
#> 2 GO:0005615(extracellular space)
#> 3 GO:0005615(extracellular space)
#> 4 GO:0005737(cytoplasm)
#> 5 <NA>
#> 6 GO:0016021(integral component of membrane)
#> molecular_function
#> 1 GO:0019904(protein domain specific binding)
#> 2 GO:0004866(endopeptidase inhibitor activity)
#> 3 GO:0004866(endopeptidase inhibitor activity)
#> 4 GO:0004813(alanine-tRNA ligase activity);GO:0005524(ATP binding);GO:0000049(tRNA binding);GO:0008270(zinc ion binding)
#> 5 GO:0003867(4-aminobutyrate transaminase activity);GO:0030170(pyridoxal phosphate binding)
#> 6 GO:0005524(ATP binding);GO:0016887(ATPase activity);GO:0042626(ATPase-coupled transmembrane transporter activity)Get help using command ?TOmicsVis::table_split or
reference page https://benben-miao.github.io/TOmicsVis/reference/table_split.html.
# Get help with command in R console.
# ?TOmicsVis::table_splitInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Table: Table merge used to merge multiple variables to on variable.
# 1. Load example datasets
data(gene_go_kegg)
head(gene_go_kegg)
#> Genes
#> 1 FN1
#> 2 14-3-3ZETA
#> 3 A1I3
#> 4 A2M
#> 5 AARS
#> 6 ABAT
#> biological_process
#> 1 GO:0003181(atrioventricular valve morphogenesis);GO:0003128(heart field specification);GO:0001756(somitogenesis)
#> 2 <NA>
#> 3 <NA>
#> 4 <NA>
#> 5 GO:0006419(alanyl-tRNA aminoacylation)
#> 6 GO:0009448(gamma-aminobutyric acid metabolic process)
#> cellular_component
#> 1 GO:0005576(extracellular region)
#> 2 <NA>
#> 3 GO:0005615(extracellular space)
#> 4 GO:0005615(extracellular space)
#> 5 GO:0005737(cytoplasm)
#> 6 <NA>
#> molecular_function
#> 1 <NA>
#> 2 GO:0019904(protein domain specific binding)
#> 3 GO:0004866(endopeptidase inhibitor activity)
#> 4 GO:0004866(endopeptidase inhibitor activity)
#> 5 GO:0004813(alanine-tRNA ligase activity);GO:0005524(ATP binding);GO:0000049(tRNA binding);GO:0008270(zinc ion binding)
#> 6 GO:0003867(4-aminobutyrate transaminase activity);GO:0030170(pyridoxal phosphate binding)
#> kegg_pathway
#> 1 ko04810(Regulation of actin cytoskeleton);ko04510(Focal adhesion);ko04151(PI3K-Akt signaling pathway);ko04512(ECM-receptor interaction)
#> 2 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko04610(Complement and coagulation cascades)
#> 5 ko00970(Aminoacyl-tRNA biosynthesis)
#> 6 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
# 2. Run function
res <- table_merge(
data = gene_go_kegg,
merge_vars = c("biological_process", "cellular_component", "molecular_function"),
new_var = "go_category",
new_value = "go_term",
na_remove = FALSE
)
head(res)
#> Genes
#> 1 FN1
#> 2 14-3-3ZETA
#> 3 A1I3
#> 4 A2M
#> 5 AARS
#> 6 ABAT
#> kegg_pathway
#> 1 ko04810(Regulation of actin cytoskeleton);ko04510(Focal adhesion);ko04151(PI3K-Akt signaling pathway);ko04512(ECM-receptor interaction)
#> 2 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko04610(Complement and coagulation cascades)
#> 5 ko00970(Aminoacyl-tRNA biosynthesis)
#> 6 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
#> go_category
#> 1 biological_process
#> 2 biological_process
#> 3 biological_process
#> 4 biological_process
#> 5 biological_process
#> 6 biological_process
#> go_term
#> 1 GO:0003181(atrioventricular valve morphogenesis);GO:0003128(heart field specification);GO:0001756(somitogenesis)
#> 2 <NA>
#> 3 <NA>
#> 4 <NA>
#> 5 GO:0006419(alanyl-tRNA aminoacylation)
#> 6 GO:0009448(gamma-aminobutyric acid metabolic process)Get help using command ?TOmicsVis::table_merge or
reference page https://benben-miao.github.io/TOmicsVis/reference/table_merge.html.
# Get help with command in R console.
# ?TOmicsVis::table_mergeInput Data: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Table: Table filter used to filter row by column condition.
# 1. Load example datasets
data(traits_sex)
head(traits_sex)
#> Value Traits Sex
#> 1 36.74 Weight Female
#> 2 38.54 Weight Female
#> 3 44.91 Weight Female
#> 4 43.53 Weight Female
#> 5 39.03 Weight Female
#> 6 26.01 Weight Female
# 2. Run function
res <- table_filter(
data = traits_sex,
Sex == "Male" & Traits == "Weight" & Value > 40
)
head(res)
#> Value Traits Sex
#> 1 48.06 Weight Male
#> 2 42.74 Weight Male
#> 3 45.25 Weight Male
#> 4 44.95 Weight Male
#> 5 43.21 Weight Male
#> 6 40.02 Weight MaleGet help using command ?TOmicsVis::table_filter or
reference page https://benben-miao.github.io/TOmicsVis/reference/table_filter.html.
# Get help with command in R console.
# ?TOmicsVis::table_filterInput Data1: Dataframe: Shared DEGs of all paired comparisons in all samples expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~: Samples).
Input Data2: Dataframe: GO and KEGG annotation of background genes (1st-col: Genes, 2nd-col: biological_process, 3rd-col: cellular_component, 4th-col: molecular_function, 5th-col: kegg_pathway).
Output Plot: Table cross used to cross search and merge results in two tables.
# 1. Load example datasets
data(gene_expression2)
head(gene_expression2)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2 LT15_3 LT12_1
#> 1 ACAA2 24.50 39.83 55.38 114.11 159.32 96.88 169.56 464.84 182.66 116.08
#> 2 ACAN 14.97 18.71 10.30 71.23 142.67 213.54 253.15 320.80 104.15 174.02
#> 3 ADH1 1.54 1.56 2.04 14.95 13.60 15.87 12.80 17.74 6.06 10.97
#> 4 AHSG 0.00 1911.99 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 ALDH2 2.07 2.86 2.54 0.85 0.49 0.47 0.42 0.13 0.26 0.00
#> 6 AP1S3 6.62 14.59 9.30 24.90 33.94 23.19 24.00 36.08 27.40 24.06
#> LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 497.29 464.48 471.43 693.62 229.77
#> 2 305.81 469.48 1291.90 991.90 966.77
#> 3 10.71 30.95 9.84 10.91 7.28
#> 4 0.00 0.00 0.00 0.00 0.00
#> 5 0.28 0.11 0.37 0.15 0.11
#> 6 38.74 34.54 62.72 41.36 28.75
data(gene_go_kegg)
head(gene_go_kegg)
#> Genes
#> 1 FN1
#> 2 14-3-3ZETA
#> 3 A1I3
#> 4 A2M
#> 5 AARS
#> 6 ABAT
#> biological_process
#> 1 GO:0003181(atrioventricular valve morphogenesis);GO:0003128(heart field specification);GO:0001756(somitogenesis)
#> 2 <NA>
#> 3 <NA>
#> 4 <NA>
#> 5 GO:0006419(alanyl-tRNA aminoacylation)
#> 6 GO:0009448(gamma-aminobutyric acid metabolic process)
#> cellular_component
#> 1 GO:0005576(extracellular region)
#> 2 <NA>
#> 3 GO:0005615(extracellular space)
#> 4 GO:0005615(extracellular space)
#> 5 GO:0005737(cytoplasm)
#> 6 <NA>
#> molecular_function
#> 1 <NA>
#> 2 GO:0019904(protein domain specific binding)
#> 3 GO:0004866(endopeptidase inhibitor activity)
#> 4 GO:0004866(endopeptidase inhibitor activity)
#> 5 GO:0004813(alanine-tRNA ligase activity);GO:0005524(ATP binding);GO:0000049(tRNA binding);GO:0008270(zinc ion binding)
#> 6 GO:0003867(4-aminobutyrate transaminase activity);GO:0030170(pyridoxal phosphate binding)
#> kegg_pathway
#> 1 ko04810(Regulation of actin cytoskeleton);ko04510(Focal adhesion);ko04151(PI3K-Akt signaling pathway);ko04512(ECM-receptor interaction)
#> 2 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko04610(Complement and coagulation cascades)
#> 5 ko00970(Aminoacyl-tRNA biosynthesis)
#> 6 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
# 2. Run function
res <- table_cross(
data1 = gene_expression2,
data2 = gene_go_kegg,
inter_var = "Genes",
left_index = TRUE,
right_index = TRUE
)
head(res)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2 LT15_3 LT12_1
#> 1 14-3-3ZETA NA NA NA NA NA NA NA NA NA NA
#> 2 A1I3 NA NA NA NA NA NA NA NA NA NA
#> 3 A2M NA NA NA NA NA NA NA NA NA NA
#> 4 AARS NA NA NA NA NA NA NA NA NA NA
#> 5 ABAT NA NA NA NA NA NA NA NA NA NA
#> 6 ABCB7 NA NA NA NA NA NA NA NA NA NA
#> LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 NA NA NA NA NA
#> 2 NA NA NA NA NA
#> 3 NA NA NA NA NA
#> 4 NA NA NA NA NA
#> 5 NA NA NA NA NA
#> 6 NA NA NA NA NA
#> biological_process
#> 1 <NA>
#> 2 <NA>
#> 3 <NA>
#> 4 GO:0006419(alanyl-tRNA aminoacylation)
#> 5 GO:0009448(gamma-aminobutyric acid metabolic process)
#> 6 <NA>
#> cellular_component
#> 1 <NA>
#> 2 GO:0005615(extracellular space)
#> 3 GO:0005615(extracellular space)
#> 4 GO:0005737(cytoplasm)
#> 5 <NA>
#> 6 GO:0016021(integral component of membrane)
#> molecular_function
#> 1 GO:0019904(protein domain specific binding)
#> 2 GO:0004866(endopeptidase inhibitor activity)
#> 3 GO:0004866(endopeptidase inhibitor activity)
#> 4 GO:0004813(alanine-tRNA ligase activity);GO:0005524(ATP binding);GO:0000049(tRNA binding);GO:0008270(zinc ion binding)
#> 5 GO:0003867(4-aminobutyrate transaminase activity);GO:0030170(pyridoxal phosphate binding)
#> 6 GO:0005524(ATP binding);GO:0016887(ATPase activity);GO:0042626(ATPase-coupled transmembrane transporter activity)
#> kegg_pathway
#> 1 ko04110(Cell cycle);ko04114(Oocyte meiosis);ko04390(Hippo signaling pathway);ko04391(Hippo signaling pathway -fly);ko04013(MAPK signaling pathway - fly);ko04151(PI3K-Akt signaling pathway);ko04212(Longevity regulating pathway - worm)
#> 2 ko04610(Complement and coagulation cascades)
#> 3 ko04610(Complement and coagulation cascades)
#> 4 ko00970(Aminoacyl-tRNA biosynthesis)
#> 5 ko00250(Alanine, aspartate and glutamate metabolism);ko00280(Valine, leucine and isoleucine degradation);ko00650(Butanoate metabolism);ko00640(Propanoate metabolism);ko00410(beta-Alanine metabolism);ko04727(GABAergic synapse)
#> 6 ko02010(ABC transporters)Get help using command ?TOmicsVis::table_cross or
reference page https://benben-miao.github.io/TOmicsVis/reference/table_cross.html.
# Get help with command in R console.
# ?TOmicsVis::table_crossThese binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
Health stats visible at Monitor.